

Key Points
Reversible complementation of FA pathway defects in human IPSCs permits derivation of isogenic mutant and control hematopoietic progenitor cells.
FANCA-deficient cells undergo accelerated erythroid differentiation because of activation of the p53-p21 axis.

Abstract
Fanconi anemia (FA) is a disorder of DNA repair that manifests as bone marrow (BM) failure. The lack of accurate murine models of FA has refocused efforts toward differentiation of patient-derived induced pluripotent stem cells (IPSCs) to hematopoietic progenitor cells (HPCs). However, an intact FA DNA repair pathway is required for efficient IPSC derivation, hindering these efforts. To overcome this barrier, we used inducible complementation of FANCA-deficient IPSCs, which permitted robust maintenance of IPSCs. Modulation of FANCA during directed differentiation to HPCs enabled the production of FANCA-deficient human HPCs that recapitulated FA genotoxicity and hematopoietic phenotypes relative to isogenic FANCA-expressing HPCs. FANCA-deficient human HPCs underwent accelerated terminal differentiation driven by activation of p53/p21. We identified growth arrest specific 6 (GAS6) as a novel target of activated p53 in FANCA-deficient HPCs and modulate GAS6 signaling to rescue hematopoiesis in FANCA-deficient cells. This study validates our strategy to derive a sustainable, highly faithful human model of FA, uncovers a mechanism of HPC exhaustion in FA, and advances toward future cell therapy in FA.
Introduction
Most patients with Fanconi anemia (FA) will develop failure of bone marrow (BM) function, typically occurring in the first decade of life. Curative therapy requires allogeneic hematopoietic stem cell transplantation, a therapy constrained by the availability of human leukocyte antigen matched donors and complications including graft-versus-host disease.1 Therefore, to benefit patients for whom stem cell transplantation is not an option, investigation has focused on the development of alternative therapies.2-4 To this end, several model systems have been engineered as platforms for mechanistic investigation of FA hematopoietic progenitor cell (HPC) dysfunction. FA is caused by mutations in at least 22 genes encoding proteins that function in the FA DNA repair pathway.5 However, knockout of single FA genes in mice typically does not produce HPC failure unless HPCs experience replicative stress, reinforcing the need for human-based systems in which to validate observations made in mouse models.6-9
The discovery of human induced pluripotent stem cell (IPSC) technology presented the opportunity for new human FA models.10 Transduction of terminally differentiated somatic cells such as skin fibroblasts or leukocytes with a defined set of transcription factors confers pluripotency and the capacity for differentiation to tissue derivatives of all 3 embryonic germ layers.11-13 Because IPSCs share these characteristics with embryonic stem cells, protocols for morphogen directed differentiation of embryonic stem cells to hematopoietic lineages can be applied to patient-derived IPSCs to model blood diseases and investigate new approaches to gene therapy or drug discovery.11,14,15 Human IPSC-based models have been used to study the BM failure disorders Diamond-Blackfan anemia and Shwachman-Diamond syndrome.16,17 However, modeling of FA has proven challenging because of the requirement for an intact FA DNA repair pathway for effective induction and maintenance of pluripotency.18-21 Rescue of FA genetic defects by introduction of a complementing cDNA in FA somatic cells can rescue reprogramming, but permanent repair of FA pathway mutations in IPSCs precludes the downstream derivation of FA-deficient HPCs for disease modeling.18,19
Here, we used a system of conditional FA pathway complementation wherein FANCA-mutated, patient-derived IPSCs bear an inducible FANCA expression cassette.22 We used these cells in a hematopoietic-directed differentiation system to derive definitive FA HPCs and isogenic control HPCs.23 FANCA-deficient, IPSC-derived HPCs show phenotypes consistent with human FA, including sensitivity to genotoxic stress and diminished clonogenicity in culture. Using this system, we found that activation of the p53/p21 axis in FANCA-deficient HPCs hinders cell cycle progression and drives terminal differentiation. We identify growth arrest specific 6 (GAS6) as a target of p53 during differentiation and show that modulation of GAS6 signaling can rescue hematopoiesis in FANCA-deficient HPCs. This system overcomes the challenges of studying FA using IPSCs and provides a renewable source of human FA HPCs and isogenic controls for further study of FA pathobiology.
Methods
Cell culture
For embryoid body (EB) formation, IPSCs were plated onto irradiated mouse embryonic fibroblast layers and cultured for 6 to 8 days in the presence of 20% knockout serum replacement (Life Technologies) with 20 ng/mL basic fibroblast growth factor (bFGF; Gibco) and 2 μg/mL doxycycline. Colonies were released by digestion with collagenase IV (Gibco) and cultured under low adherence conditions to form EBs.23 On day 8, EBs were dissociated, and CD34+ hemogenic endothelium (HE) was affinity purified using anti-human CD34 microbeads (Miltenyi Biotech) and seeded on Matrigel-coated plates.
For endothelial to hematopoietic transition (EHT) culture, cells were cultured in EB medium supplemented with 5 ng/mL bFGF, 15 ng/mL vascular endothelial growth factor, 10 ng/mL interleukin 6 (IL-6), 5 ng/mL IL-11, 25 ng/mL insulin-like growth factor 1, 50 ng/mL stem cell factor, 2 U/mL erythropoietin, 10 ng/mL bone morphogenetic protein 4 (BMP4), 30 ng/mL thrombopoietin, 10 ng/mL Flt3 ligand, and 30 ng/mL IL-3 for 8 days in 5% oxygen (EHT culture). Where indicated, doxycycline was either removed or maintained in culture starting at day 0 of EHT culture. For erythroid differentiation, hematopoietic cells were harvested at day 8 of EHT culture and further cultured in EHT medium. For neutrophil differentiation, EB formation and EHT were performed in the absence of EPO, and nonadherent cells were cultured with 50 ng/mL stem cell factor and 50 ng/mL granulocyte colony-stimulating factor for 4 days, at which time they were analyzed by flow cytometry. Recombinant human GAS6 was used at 800 ng/mL where indicated. For StemDiff culture, IPSCs were cultured with StemDiff reagents per the manufacturer’s protocol (StemCell Technologies).
For methylcellulose culture, HPCs were collected at day 5 or 8 of EHT. A total of 1000 to 10 000 viable cells were suspended in 1.5 mL MethoCult SF H4636 (Stem Cell Technologies) supplemented with 50 ng/mL IL-6, 50 ng/mL thrombopoietin, and 50 ng/mL Flt3 ligand. Colonies were enumerated by blinded scoring at day 14. Where indicated, nutlin-3a at a final concentration of 1 μM was added.
Gene editing
IPSCs were transfected using the 4-D nucleofector platform (Lonza) with crRNA/Cas9 ribonucleoprotein complexes (IDT Technologies) and assembled according to the manufacturer’s protocol. Transfected cells were enriched for those with biallelic TP53 inactivation by culture with nutlin-3a at a concentration of 5 μM for 7 days. Frequency of insertions and deletions was measured by deep sequencing of a polymerase chain reaction (PCR) product amplified with primers flanking the crRNA binding site.
Statistics
Analyses were performed using R, Microsoft Excel, and Graphpad Prism. An unpaired Student t test was used with a significance cutoff of 0.05 unless otherwise stated. Results are presented as mean ± standard error of mean (SEM) unless otherwise stated.
Flow cytometry
The antibodies used in this study were all against human antigens: anti-CD71 allophycocyanin (APC), anti-GLYA PE-Cy7, anti-CD49d phycoerythrin (PE), anti-CD45 APC, anti-CD45 fluorescein isothiocyanate, anti-Ki67 APC, anti-CD34 PE-Cy7 (all from BD Biosciences), anti-Band 3 fluorescein isothiocyanate (American Research Products), anti-γH2AX (EMD Millipore), and anti-FANCD2 (Novus). Annexin V APC was used (BD Pharmingen).
Cell cycle analysis
Cells were collected in phosphate-buffered saline and fixed with 70% ethanol. FxCycle PI/RNase staining solution (Molecular Probes/Life Technologies) or a combination of 4′,6-diamidino-2-phenylindole (DAPI) counterstain with Ki67 immunostaining was used for flow cytometry analysis.
RNA sequencing
HPCs were collected at day 8 of EHT culture and resuspended in Trizol (Thermo Fisher Scientific) followed by RNA purification and on-column DNAase treatment using the RNeasy Kit (Qiagen). Three biologic replicates for each condition were included. Low input library preparation was performed by the Molecular Biology Core Facility at the Dana-Farber Cancer Institute (Boston, MA). Libraries were sequenced using the Illumina NextSeq 500 Single-End 75 bp platform with expected 400 million read count.
Quantitative PCR
Whole RNA was used to synthesize cDNA using the miScript Reverse Transcription Kit (Qiagen). Primer sequences were as follows—FANCA forward: CTTCCGAGAGGTGTTGAAAGA; FANCA reverse: GAAGTCCTGCCGTTCAGTATC; ACTB forward: GGATCAGCAAGCAGGAGTATG; ACTB reverse: AGAAAGGGTGTAACGCAACTAA; CDKN1A forward: CTGGGGATGTCCGTCAGAAC; CDKN1A reverse: CATTAGCGCATCACAGTCGC; GAS6 forward: ACCTGTGAGGACATCTTGCC; GAS6 reverse: GGGTCAAAGGTCCGGAAGTC.
Teratoma assay
One million IPSCs were harvested from Matrigel cultures. Cells were resuspended in a 1:1 mixture of DMEM/F12 medium and Matrigel and injected subcutaneously into NOD.Cg-PrkdcscidIL2rgtmlWjl/SzJ (NOD/SCID/IL2rγnull; NSG) mice. After 10 to 12 weeks, teratomas were harvested, fixed, embedded, sectioned, and stained for analysis.
Immunofluorescence
For the p53 immunostain, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. The DO-7 mouse monoclonal antibody was used (Cell Signaling Technologies) with an Alexa Fluor 594 goat anti-mouse secondary antibody. To costain for FANCD2/γH2AX, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 followed by immunostaining for FANCD2 (Novus) and γH2AX (EMD Millipore). For quantification of nuclear p53, images were acquired using a 40× lens on a CV7000 CellVoyager Measurement System (version R1.17.06). A script (run on Fiji, V2.0.0-rc-68/1.52g) was used to further process and analyze the images.
Patient cells
FA patient BM aspirates were collected on a human subjects study approved by the Institutional Review Board at Boston Children’s Hospital, after informed consent was obtained. This study was performed in accordance with the Declaration of Helsinki. Normal healthy BM mononuclear cells were purchased from AllCells.
Enzyme linked immunosorbent assay
Human GAS6 enzyme linked immunosorbent assay (ELISA) was performed using the Duoset ELISA Kit (R&D Systems).
Results
Directed differentiation of FA IPSCs to HPCs
We used human patient-derived, FANCA homozygous mutant IPSC lines that harbor a stably integrated doxycycline-inducible transgene encoding the wild-type FANCA open reading frame for inducible and reversible expression.22 For this study, we used the FA-A#1 and FA-A#2 lines that have previously been characterized as pluripotent22 and a subclone of FA-A#2 that was verified as pluripotent and euploid in our hands (supplemental Table 1; supplemental Figure 1). For subsequent experiments, unless otherwise indicated, we pooled results from experiments using all 3 of these IPSC lines to limit effects of variability in differentiation potential between lines attributable to background genetic or clonal variation.24-26
We adapted previously described protocols of EB-based directed differentiation to generate human HE (Figure 1A).23,27 For unperturbed directed differentiation and specification of HE, we maintained EBs with doxycycline throughout this 8-day phase of differentiation (Figure 1A). FA IPSCs robustly formed EBs (Figure 1B). We dissociated day 8 EBs and confirmed that we could detect cells expressing a CD34+GLYA−CD43−KDR+ HE immunophenotype (Figure 1C).23 We isolated HE from dissociated EBs based on surface CD34 and found that adherent HE showed the expected morphology (Figure 1D).
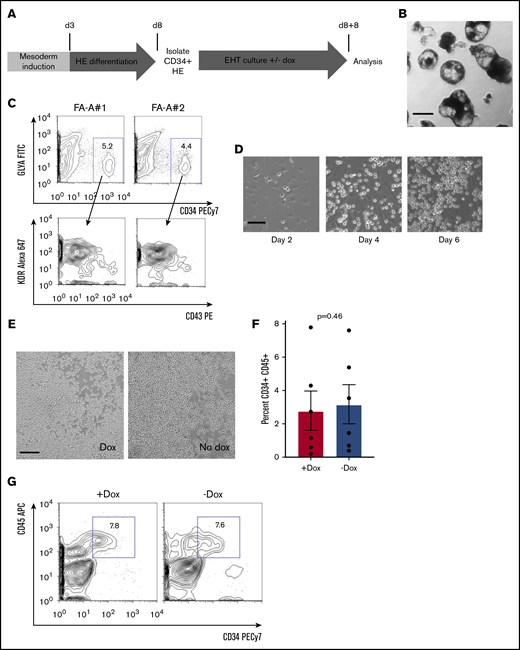
Hematopoietic differentiation of doxycycline inducible human FANCA patient IPSCs. (A) Schema for the differentiation of human IPSCs to HPCs adapted from published protocols.23,27 The first 8-day period involves morphogen-directed specification of hemogenic endothelium within EBs (see "Methods"). This period of culture incorporates 2 phases: BMP-4 and bFGF-mediated mesoderm induction, followed by HE differentiation driven by vascular endothelial growth factor, IL-11, insulin-like growth factor 1, and hematopoietic cytokines. At day 8, EBs were dissociated, and HE was isolated based on CD34+ selection. HE was plated on a 2-dimensional Matrigel substratum for an 8-day EHT culture in the presence of hematopoietic cytokines with or without doxycycline to modulate FANCA expression. At the end of the 8-day EHT culture, round floating HPCs were harvested for analysis in assays of clonogenicity and hematopoietic differentiation. (B) Representative image of day 8 EBs before dissociation (scale bar, 200 μm). (C) The entire cellular composition of dissociated day 8 EBs derived from the indicated human IPSC lines was stained with the indicated antibodies and definitive HE (CD34+GLYA−KDR+CD43−) was analyzed by flow cytometry gated on viable singlet cells. (D) Representative images of cultured HE at the indicated time points during EHT culture (scale bar, 50 μm). (E) Representative images of day 8 EHT culture in the presence or absence of doxycycline (scale bar, 100 μm). (F-G) Nonadherent, round cells were harvested at day 8 EHT and analyzed for expression of the indicated surface markers of HPCs by flow cytometry. Percent CD34+CD45+ HPCs within the nonadherent population was quantified over 6 independent experiments (results presented as mean ± SEM, analyzed by a paired Student t test).
Hematopoietic differentiation of doxycycline inducible human FANCA patient IPSCs. (A) Schema for the differentiation of human IPSCs to HPCs adapted from published protocols.23,27 The first 8-day period involves morphogen-directed specification of hemogenic endothelium within EBs (see "Methods"). This period of culture incorporates 2 phases: BMP-4 and bFGF-mediated mesoderm induction, followed by HE differentiation driven by vascular endothelial growth factor, IL-11, insulin-like growth factor 1, and hematopoietic cytokines. At day 8, EBs were dissociated, and HE was isolated based on CD34+ selection. HE was plated on a 2-dimensional Matrigel substratum for an 8-day EHT culture in the presence of hematopoietic cytokines with or without doxycycline to modulate FANCA expression. At the end of the 8-day EHT culture, round floating HPCs were harvested for analysis in assays of clonogenicity and hematopoietic differentiation. (B) Representative image of day 8 EBs before dissociation (scale bar, 200 μm). (C) The entire cellular composition of dissociated day 8 EBs derived from the indicated human IPSC lines was stained with the indicated antibodies and definitive HE (CD34+GLYA−KDR+CD43−) was analyzed by flow cytometry gated on viable singlet cells. (D) Representative images of cultured HE at the indicated time points during EHT culture (scale bar, 50 μm). (E) Representative images of day 8 EHT culture in the presence or absence of doxycycline (scale bar, 100 μm). (F-G) Nonadherent, round cells were harvested at day 8 EHT and analyzed for expression of the indicated surface markers of HPCs by flow cytometry. Percent CD34+CD45+ HPCs within the nonadherent population was quantified over 6 independent experiments (results presented as mean ± SEM, analyzed by a paired Student t test).
After purification of HE, we initiated EHT culture (Figure 1A). At the initiation of EHT, doxycycline was either removed or maintained for the subsequent 8 days to generate FANCA-deficient or FANCA-expressing HPCs, respectively (Figure 1A). During EHT, we observed the progressive budding of round, nonadherent human HPCs from the adherent HE layer (Figure 1D), which was not affected by the presence or absence of doxycycline (Figure 1E). Moreover, at the completion of EHT, we detected equivalent proportions of cells expressing the HPC markers CD45 and CD34, further supportive of unimpaired EHT in the absence of doxycycline (Figure 1F-G). As an independent method, we used the StemDiff system, again observing no difference in HPC emergence (Stem Cell Technologies; supplemental Figure 2).
We next examined the status of the FA pathway after EHT. We found that in HPCs generated without doxycycline contained approximately 20% FANCA mRNA compared with HPCs generated with doxycycline (Figure 2A). To test the functionality of the FA pathway in response to genotoxic stress, we exposed HPCs to mitomycin C, finding that FANCA-deficient cells were unable to localize FANCD2 at sites of DNA damage marked by γH2AX as observed in FANCA-expressing cells (Figure 2B-C). These results indicate the absence of functional FANCA expression in HPCs emerging in the absence of doxycycline.
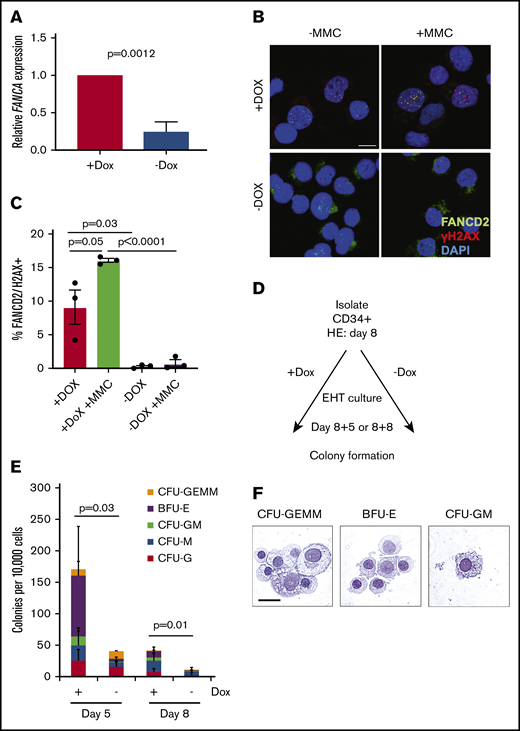
Analysis of FANCA-deficient and FANCA-expressing IPSC-derived hematopoietic cells. (A) Total RNA was isolated from the nonadherent cells at day 8 EHT, and expression of FANCA was analyzed by quantitative PCR (n = 3 biologic replicates; results presented as mean ± SEM, analyzed by a Student t test). (B-C) Nonadherent cells were isolated at day 8 of EHT after culture with or without doxycycline and then cultured with mitomycin C (20 ng/mL) for 18 hours, at which time the cells were fixed and immunostained for FANCD2 and γH2AX and visualized by confocal microscopy (scale bar, 10 μm). Results are compiled from 3 separate lines, and the proportion of cells with double-positive foci presented as mean ± SEM, analyzed by a paired Student t test. (D-E) A total of 10 000 nonadherent cells collected from day 5 or day 8 EHT cultures were embedded into methylcellulose with hematopoietic growth factors. Doxycycline was either maintained in these assays or withheld consistent with EHT culture conditions. After 14 days, colonies were scored morphologically, and the results were quantified (n = 7 biologic replicates for day 5 and n = 10 biologic replicates day 8 across 3 cell lines; total colony numbers analyzed by a paired Student t test, results are presented as mean proportions of each colony type ± standard deviation). (F) Hematopoietic colonies were isolated at day 14 from cultures with doxycycline and spun onto slides when the cells were stained with May-Grünwald-Giemsa and analyzed by light microscopy (scale bar, 10 μm).
Analysis of FANCA-deficient and FANCA-expressing IPSC-derived hematopoietic cells. (A) Total RNA was isolated from the nonadherent cells at day 8 EHT, and expression of FANCA was analyzed by quantitative PCR (n = 3 biologic replicates; results presented as mean ± SEM, analyzed by a Student t test). (B-C) Nonadherent cells were isolated at day 8 of EHT after culture with or without doxycycline and then cultured with mitomycin C (20 ng/mL) for 18 hours, at which time the cells were fixed and immunostained for FANCD2 and γH2AX and visualized by confocal microscopy (scale bar, 10 μm). Results are compiled from 3 separate lines, and the proportion of cells with double-positive foci presented as mean ± SEM, analyzed by a paired Student t test. (D-E) A total of 10 000 nonadherent cells collected from day 5 or day 8 EHT cultures were embedded into methylcellulose with hematopoietic growth factors. Doxycycline was either maintained in these assays or withheld consistent with EHT culture conditions. After 14 days, colonies were scored morphologically, and the results were quantified (n = 7 biologic replicates for day 5 and n = 10 biologic replicates day 8 across 3 cell lines; total colony numbers analyzed by a paired Student t test, results are presented as mean proportions of each colony type ± standard deviation). (F) Hematopoietic colonies were isolated at day 14 from cultures with doxycycline and spun onto slides when the cells were stained with May-Grünwald-Giemsa and analyzed by light microscopy (scale bar, 10 μm).
Hematopoietic potential of FA HPCs derived from IPSCs
Next, to determine whether our system recapitulates the hallmarks of HPC dysfunction in FA, we examined the hematopoietic capacity of FANCA-deficient HPCs.28 We cultured IPSC-derived HPCs either with or without doxycycline to either maintain or deactivate the inducible FANCA transgene, respectively, during assays of hematopoietic function (Figure 2D). In colony formation assays, FANCA-deficient HPCs collected at either day 5 or day 8 EHT showed significantly diminished clonogenicity relative to control FANCA-expressing cells after 14 days (Figure 2E-F). As expected, colony-forming capacity diminished from day 5 to day 8 because of differentiation of HPCs in culture. We observed similarly impaired clonogenicity of FANCA-deficient HPCs derived independently via the StemDiff system and in primary mononuclear cells isolated from FA patients compared with normal healthy control donors (supplemental Figure 2). These results indicate that FANCA deficiency in HPCs derived from IPSCs impairs hematopoietic colony formation, recapitulating a known phenotype of HPCs from the BM of human FA patients.28
Transcriptional profile of FANCA-deficient HPCs
Next, we performed RNA sequencing to evaluate the effect of FANCA deficiency on the transcriptional profile of human IPSC-derived HPCs. Using CellNet, a cell state prediction algorithm,29 we found that all specimens included in this analysis classified as HPCs without residual signatures of endothelial cells, consistent with unimpaired EHT (Figure 3A). We found 422 transcripts differentially expressed between FANCA-expressing and FANCA-deficient IPSC-derived HPCs at a significance cutoff of 0.05. We used the Molecular Signatures Database to probe signatures modulated by FANCA.30 Using gene set enrichment analysis (GSEA), we observed enrichment of terms related to mitochondrial respiration in FANCA-expressing cells, consistent with a reported role of the FA pathway in regulation of mitochondrial function (Figure 3B).31,32 Furthermore, we observed signatures consistent with active oxidative phosphorylation, ribosome biogenesis, and translation (Figure 3B; supplemental Figure 3). We found that FANCA-expressing HPCs more strongly maintained a normal human HPC signature relative to FANCA-deficient HPCs30,33 (Figure 3C). In FANCA-expressing cells, we also observed enrichment of signatures related to regulation of the G2/M checkpoint and DNA repair, consistent with the known biology of FA (supplemental Figure 3).34,35 Moreover, we found signatures associated with the acute inflammatory response including IL-1 and interferon-β production enriched and Toll-like receptors in FANCA-deficient cells as has been described previously in mice36,37 (supplemental Figure 4). We did not observe signatures suggestive of perturbed transforming growth factor-β signaling in FANCA-deficient cells and observed partial overlap with transcripts altered in Fancd2−/− mouse hematopoietic stem and progenitor cells vs wild type (supplemental Figure 4).2,38,39 Interestingly, we found enrichment for transcripts associated with heme metabolism and terminal erythropoiesis in FANCA-deficient HPCs, suggestive of an erythroid committed state in these cells (Figure 3D-E). Together, these findings show that our FANCA-deficient HPCs bear transcriptional signatures consistent with perturbation of HPC state, recapitulation of known transcriptional signatures associated with FA, as well as a novel signature related to terminal erythropoiesis.
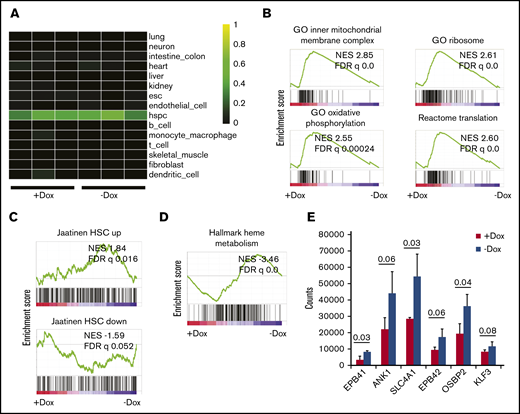
Analysis of gene expression by RNA sequencing. (A) Heatmap showing gene regulatory network enrichment scores as analyzed by CellNet29 for FANCA-expressing (Dox) and FANCA-deficient (No dox) hematopoietic cells from day 8 EHT culture. Individual profiles for each of 3 biologic replicates from both cell types are shown. (B) GSEA was used to query the Molecular Signatures Database,30 and relevant terms enriched in FANCA-expressing cells relative to FANCA-deficient cells are presented. In all GSEA analyses, a preranked transcript list was used that was compiled from 3 biologic replicates of sequencing for both FANCA-expressing and FANCA-deficient hematopoietic cells. (C) GSEA was used to query gene sets enriched in human umbilical cord blood HSCs relative to non-HSCs.33 (D) GSEA was used to query the Molecular Signatures Database, and terms enriched in FANCA-deficient cells relative to FANCA-expressing cells are presented. (E) Expression of erythroid genes in FANCA-expressing vs FANCA-deficient hematopoietic cells (analysis with a Student t test, n = 3 biologic replicates, results presented as mean ± standard deviation; P values are shown).
Analysis of gene expression by RNA sequencing. (A) Heatmap showing gene regulatory network enrichment scores as analyzed by CellNet29 for FANCA-expressing (Dox) and FANCA-deficient (No dox) hematopoietic cells from day 8 EHT culture. Individual profiles for each of 3 biologic replicates from both cell types are shown. (B) GSEA was used to query the Molecular Signatures Database,30 and relevant terms enriched in FANCA-expressing cells relative to FANCA-deficient cells are presented. In all GSEA analyses, a preranked transcript list was used that was compiled from 3 biologic replicates of sequencing for both FANCA-expressing and FANCA-deficient hematopoietic cells. (C) GSEA was used to query gene sets enriched in human umbilical cord blood HSCs relative to non-HSCs.33 (D) GSEA was used to query the Molecular Signatures Database, and terms enriched in FANCA-deficient cells relative to FANCA-expressing cells are presented. (E) Expression of erythroid genes in FANCA-expressing vs FANCA-deficient hematopoietic cells (analysis with a Student t test, n = 3 biologic replicates, results presented as mean ± standard deviation; P values are shown).
Accelerated terminal differentiation of FANCA-deficient HPCs
On further investigation of the pro-erythroid signature observed in FANCA-deficient HPCs, we found that these cells expressed higher levels of the downstream erythropoietin signaling components JAK2 and STAT5B, as well as the erythropoietin targets BCL2L1 and SOCS3 (supplemental Figure 3).40 Therefore, we next examined erythropoietin-dependent differentiation.17,41 Within the GLYA+ fraction containing committed erythroid cells, stepwise erythroid differentiation can be examined including proerythroblasts (region I; CD49d+Band 3−), early basophilic erythroblasts (II; CD49d+Band 3−low), late basophilic erythroblasts (III; CD49d+Band 3+), polychromatophilic erythroblasts (IV; CD49d−low Band 3+), and orthochromatophilic erythroblasts (V; CD49d−Band 3+; supplemental Figure 5).42 Although both FANCA-expressing and -deficient cells showed efficient early erythroid commitment with erythroid-induced acquisition of CD71 and GLYA expression (Figure 4A), we found that FANCA-deficient HPCs showed accelerated terminal erythroid differentiation (Figure 4B-D). To determine whether this phenotype was specific to erythroid cells, we also tested granulocyte colony-stimulating factor–driven myeloid differentiation, observing an effect suggestive of acceleration in FANCA deficient cells (supplemental Figure 6). Together, these findings show that FANCA deficiency predisposes HPCs to terminal differentiation.
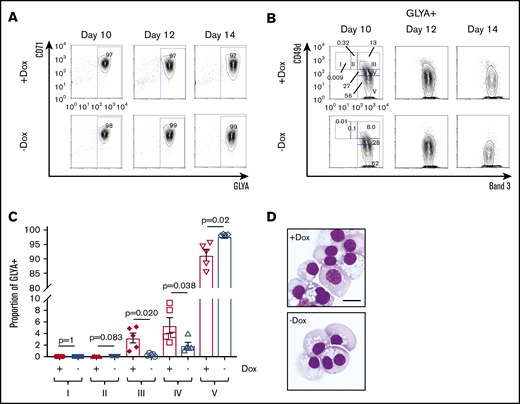
Erythroid differentiation of FANCA-expressing or -deficient IPSC-derived hematopoietic cells. (A) Day 8 nonadherent cells from EHT culture were cultured in a cytokine cocktail containing erythropoietin for the indicated periods of time after the initiation of EHT culture, when erythroid differentiation was measured by flow cytometry for CD71 and GLYA expression. (B) Within the GLYA+ gate, cell surface expression of CD49d and Band 3 was measured by flow cytometry. Stages of erythroid differentiation were quantified by gating as described previously.42 (C) Stages of erythroid differentiation were quantified at day 14, and each stage was compared by a Student t test (n = 5 biologic replicates, results presented as mean ± SEM). (D) Representative morphology of day 14 erythroid differentiation culture (scale bar, 10 μm).
Erythroid differentiation of FANCA-expressing or -deficient IPSC-derived hematopoietic cells. (A) Day 8 nonadherent cells from EHT culture were cultured in a cytokine cocktail containing erythropoietin for the indicated periods of time after the initiation of EHT culture, when erythroid differentiation was measured by flow cytometry for CD71 and GLYA expression. (B) Within the GLYA+ gate, cell surface expression of CD49d and Band 3 was measured by flow cytometry. Stages of erythroid differentiation were quantified by gating as described previously.42 (C) Stages of erythroid differentiation were quantified at day 14, and each stage was compared by a Student t test (n = 5 biologic replicates, results presented as mean ± SEM). (D) Representative morphology of day 14 erythroid differentiation culture (scale bar, 10 μm).
Impaired cell cycling promotes differentiation of FANCA-deficient HPCs
Given the known role of the FA pathway in regulation of the cell cycle and the importance of cycling in differentiation,35,43,44 we next examined the cell cycle. FANCA-deficient HPCs showed a significantly reduced proportion of cells in G1-phase and a trend toward fewer cells in S/G2/M-phases consistent with less proliferation (Figure 5A-B). We also observed higher expression of the CDKN1A transcript encoding the G1/S phase checkpoint regulator p21 in FANCA-deficient cells (Figure 5C). Consistent with this finding, we observed increased nuclear localization of p53 protein, increased p53 protein levels, and loss of an E2F transcriptional signature in FANCA-deficient cells (Figure 5D-G). Increased p53 pathway activation was associated with slightly increased apoptosis in FANCA-deficient cells (supplemental Figure 8).
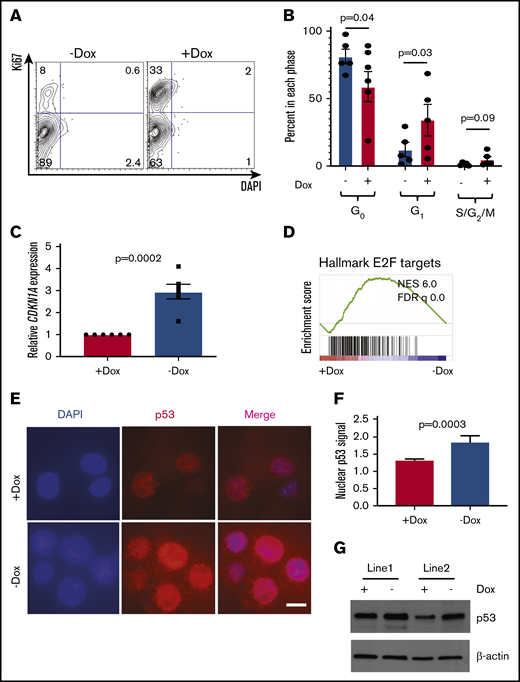
Cell cycle and apoptosis analysis in FANCA-expressing and -deficient IPSC-derived hematopoietic cells. (A-B) HPCs were isolated after the completion of EHT culture, at which time they were isolated and stained with DAPI and Ki67 to quantify the cell cycle. The percentage of cells in each phase of the cell cycle was quantified (n = 5 biologic replicates; each phase analyzed by a paired Student t test, results are presented as mean ± SEM). (C) Expression level of the CDKN1A transcript was measured by quantitative PCR and expression normalized to β-actin mRNA. Expression was compared by Student t test with 6 biologic replicates. (D) GSEA analysis of RNA sequencing data. (E-F) Subcellular localization of p53 was analyzed by immunofluorescence staining and nuclear fluorescence signal quantified normalized to DAPI signal (scale bar, 10 μm; results aggregated from 3 biologic replicates, compared by a Student t test, results presented as mean ± SEM; n = 240 Dox cells and 111 No dox cells quantified). (G) p53 protein was measured in day 8 EHT HPCs from 2 independent cell lines under the indicated conditions.
Cell cycle and apoptosis analysis in FANCA-expressing and -deficient IPSC-derived hematopoietic cells. (A-B) HPCs were isolated after the completion of EHT culture, at which time they were isolated and stained with DAPI and Ki67 to quantify the cell cycle. The percentage of cells in each phase of the cell cycle was quantified (n = 5 biologic replicates; each phase analyzed by a paired Student t test, results are presented as mean ± SEM). (C) Expression level of the CDKN1A transcript was measured by quantitative PCR and expression normalized to β-actin mRNA. Expression was compared by Student t test with 6 biologic replicates. (D) GSEA analysis of RNA sequencing data. (E-F) Subcellular localization of p53 was analyzed by immunofluorescence staining and nuclear fluorescence signal quantified normalized to DAPI signal (scale bar, 10 μm; results aggregated from 3 biologic replicates, compared by a Student t test, results presented as mean ± SEM; n = 240 Dox cells and 111 No dox cells quantified). (G) p53 protein was measured in day 8 EHT HPCs from 2 independent cell lines under the indicated conditions.
We next examined the connection between FANCA, p53, and differentiation. Erythroid differentiation is triggered by GATA1-dependent activation of p21-driven cell cycle arrest rather than apoptosis.43-45 We found that cell cycling diminishes with progression through erythroid differentiation (supplemental Figure 5). We therefore hypothesized that the enhanced p53/p21 activity in FANCA-deficient HPCs augments physiologic expression of p21 to drive erythroid differentiation. To test this, we treated FANCA-expressing cells with nutlin-3a, an inhibitor of MDM2-driven p53 degradation. Nutlin-3a induced p21 mRNA expression, impaired colony formation, and accelerated erythroid differentiation in FANCA-expressing HPCs (supplemental Figure 7). Thus, activation of p53 is sufficient to augment erythropoietin-induced erythroid differentiation at the expense of clonogenicity.
To further verify this model, we used gene editing to ablate the TP53 gene in IPSCs followed by treatment of IPSCs with nutlin-3a to increase inserted/deleted allele frequency (supplemental Figure 8). Edited cells were deficient in TP53 transcripts and could not recruit CDKN1A in response to irradiation as expected based on disruption of the TP53 locus (Figure 6A-B). FANCA-deficient, p53-disrupted HPCs showed increased cycling relative to unedited FANCA-deficient HPCs (Figure 6C-D). p53 deficiency rescued the impaired clonogenicity of FANCA-deficient HPCs (Figure 6E). Moreover, p53 deficiency in FANCA-deficient cells slowed progression of erythroid differentiation (Figure 6F-G), establishing p53 as an effector of aberrant HPC differentiation in FA.
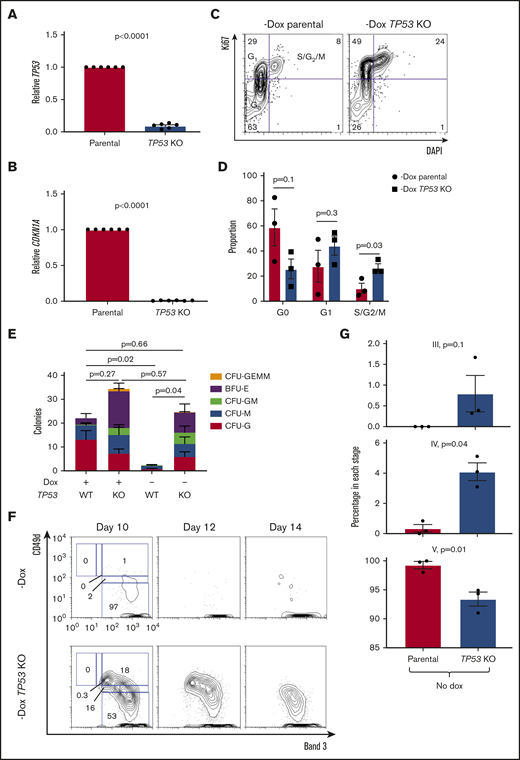
Disabling p53 rescues clonogenesis and slows differentiation in FANCA-deficient cells. (A-B) The indicated human IPSC lines were exposed to 1-Gy ionizing radiation, and expression of TP53 and CDKN1A was analyzed 4 hours later by quantitative PCR. Results are aggregated from 2 independent experiments and analyzed by a Student t test. (C-D) FANCA-deficient HPCs generated in the absence of doxycycline with or without TP53 disruption were stained for Ki67 and DAPI to analyze cell cycle status. Results are aggregated from 3 parental IPSC lines and compiled over 2 independent experiments and analyzed by a Student t test. (E) The indicated HPCs isolated at day 8 of EHT were plated in methylcellulose in the presence of hematopoietic cytokines (10 000 cells/assay), and 14 days later colonies were scored (n = 4 biologic replicates over 3 independent experiments, and total colony numbers were compared by a Student t test, results are presented as mean ± SEM for each aggregated colony type). (F-G) The indicated cell lines were cultured under pro-erythroid differentiation conditions and analyzed by flow cytometry at the indicated time points. CD49d/Band 3 plots are from the GLYA+ population of the differentiating culture. Results are aggregated over 3 independent experiments and compared by a Student t test.
Disabling p53 rescues clonogenesis and slows differentiation in FANCA-deficient cells. (A-B) The indicated human IPSC lines were exposed to 1-Gy ionizing radiation, and expression of TP53 and CDKN1A was analyzed 4 hours later by quantitative PCR. Results are aggregated from 2 independent experiments and analyzed by a Student t test. (C-D) FANCA-deficient HPCs generated in the absence of doxycycline with or without TP53 disruption were stained for Ki67 and DAPI to analyze cell cycle status. Results are aggregated from 3 parental IPSC lines and compiled over 2 independent experiments and analyzed by a Student t test. (E) The indicated HPCs isolated at day 8 of EHT were plated in methylcellulose in the presence of hematopoietic cytokines (10 000 cells/assay), and 14 days later colonies were scored (n = 4 biologic replicates over 3 independent experiments, and total colony numbers were compared by a Student t test, results are presented as mean ± SEM for each aggregated colony type). (F-G) The indicated cell lines were cultured under pro-erythroid differentiation conditions and analyzed by flow cytometry at the indicated time points. CD49d/Band 3 plots are from the GLYA+ population of the differentiating culture. Results are aggregated over 3 independent experiments and compared by a Student t test.
GAS6 is a p53 target modulating HPC function
To further investigate the role of p53 in regulating HPC function, we used K562 erythroleukemia cells either deficient for p53 or where the endogenous TP53 locus was repaired using genome editing.46 We exposed these cells to hemin to induce erythroid differentiation.47 We then performed chromatin immunoprecipitation with sequencing for p53, finding that p53 bound the GAS6 locus (Figure 7A). GAS6 is a known regulator of erythropoiesis.48 We found that GAS6 expression was increased in FANCA-deficient HPCs relative to controls at the RNA and protein levels (Figure 7B-C). Nutlin-3a increased GAS6 expression, consistent with GAS6 as a p53 target gene (Figure 7D). We initially hypothesized that p53-driven GAS6 synthesis would negatively regulate HPC function by signaling through its TYRO, AXL, and MER (TAM) receptors. We therefore inhibited TAM signaling in FANCA-deficient HPC by using the TAM receptor inhibitor BMS-777607 and activated TAM receptors in FANCA-expressing HPCs by exposing them to exogenous GAS6. We found that inhibition of TAM receptors did not rescue clonogenesis in FANCA-deficient cells, whereas exogenous GAS6 modestly increased HPC function in FANCA-expressing cells (Figure 7E).
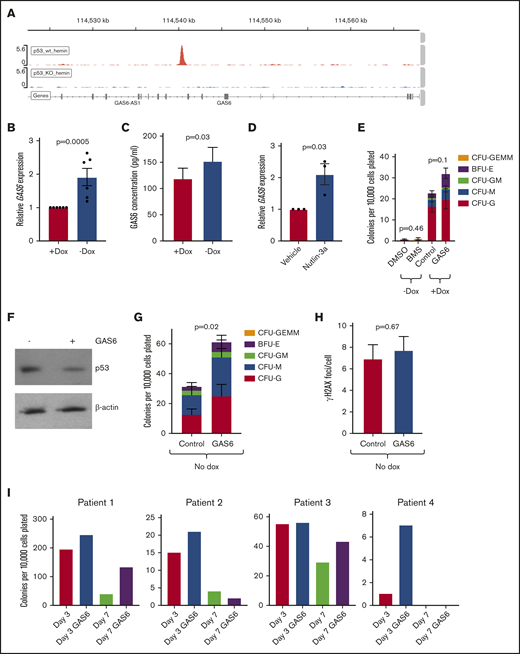
GAS6 is a p53 target in HPCs. (A) p53 wild-type (wt) or knockout (KO) K562 cells were treated with hemin to induce erythroid differentiation for 4 days, at which time chromatin immunoprecipitation for p53 was performed, with peaks at the human GAS6 locus shown. (B) FANCA-expressing or deficient HPCs generated by EHT in the presence of absence of doxycycline, respectively, were collected, and expression of GAS6 was analyzed by quantitative PCR (n = 6 biologic replicates results). (C) Conditioned culture supernatant of day 8 EHT cells was used for ELISA measurement of GAS6 protein (n = 13 biologic replicates compiled over 2 independent ELISA experiments). (D) FANCA-expressing HPCs were treated with or without nutlin-3a and expression of GAS6 measured by quantitative PCR (n = 3 biologic replicates results presented as mean ± SEM and compared by a Student t test with P value shown). (E) The indicated cells were treated either with BMS-777607 or GAS6 during EHT and then used in a colony formation assay (n = 7 biologic replicates over 3 experiments). (F) p53-repaired K562 cells were cultured serum free for 24 hours during which time they were treated with GAS6, and protein was collected for western blot analysis. (G) FANCA-deficient HPCs generated in the absence of doxycycline either in the presence of absence of recombinant GAS6 were harvested at day 5 of EHT and plated in methylcellulose medium, where colony formation was quantified after 14 days (n = 9 biologic replicates including 3 cell lines over 5 independent experiments). (H) Day 8 EHT cells generated without doxycycline and with or without GAS6 were exposed to mitomycin C and γH2AX-positive foci per cell quantified (n = 3 cell lines included and pooled in the overall analysis). (I) Primary FA patient bone marrow mononuclear cells were treated with or without GAS6 for either 3 or 7 days and then used in a colony formation assay, where colonies were quantified at day 14. All results presented as mean ± SEM and compared by a Student t test with P value shown.
GAS6 is a p53 target in HPCs. (A) p53 wild-type (wt) or knockout (KO) K562 cells were treated with hemin to induce erythroid differentiation for 4 days, at which time chromatin immunoprecipitation for p53 was performed, with peaks at the human GAS6 locus shown. (B) FANCA-expressing or deficient HPCs generated by EHT in the presence of absence of doxycycline, respectively, were collected, and expression of GAS6 was analyzed by quantitative PCR (n = 6 biologic replicates results). (C) Conditioned culture supernatant of day 8 EHT cells was used for ELISA measurement of GAS6 protein (n = 13 biologic replicates compiled over 2 independent ELISA experiments). (D) FANCA-expressing HPCs were treated with or without nutlin-3a and expression of GAS6 measured by quantitative PCR (n = 3 biologic replicates results presented as mean ± SEM and compared by a Student t test with P value shown). (E) The indicated cells were treated either with BMS-777607 or GAS6 during EHT and then used in a colony formation assay (n = 7 biologic replicates over 3 experiments). (F) p53-repaired K562 cells were cultured serum free for 24 hours during which time they were treated with GAS6, and protein was collected for western blot analysis. (G) FANCA-deficient HPCs generated in the absence of doxycycline either in the presence of absence of recombinant GAS6 were harvested at day 5 of EHT and plated in methylcellulose medium, where colony formation was quantified after 14 days (n = 9 biologic replicates including 3 cell lines over 5 independent experiments). (H) Day 8 EHT cells generated without doxycycline and with or without GAS6 were exposed to mitomycin C and γH2AX-positive foci per cell quantified (n = 3 cell lines included and pooled in the overall analysis). (I) Primary FA patient bone marrow mononuclear cells were treated with or without GAS6 for either 3 or 7 days and then used in a colony formation assay, where colonies were quantified at day 14. All results presented as mean ± SEM and compared by a Student t test with P value shown.
Given that GAS6 is a prosurvival growth factor, we next hypothesized that p53-induced GAS6 induction acts as a feedback mechanism that could be potentiated to rescue FANCA-deficient HPC progenitor function by repressing p53.49 We found that in p53-repaired K562 cells, GAS6 could decrease p53 protein levels (Figure 7F). Therefore, we treated FANCA-deficient HPCs with or without recombinant human GAS6, finding that GAS6 treatment rescued HPC clonogenesis (Figure 7G). GAS6 did not diminish DNA damage as marked by formation of γH2AX (Figure 7H). Treatment of primary FA patient hematopoietic stem and progenitor cells could improve clonogenesis in all 4 patients tested in at least 1 of the GAS6 treatment durations (Figure 7I). These results identify GAS6 is a p53 target that can be modulated to improve FA HPC function.
Discussion
In this study, we used an inducible system to complement FANCA mutations in patient-derived IPSCs to maintain the function of the FA pathway while cells are in the pluripotent state.22 By either removing or maintaining doxycycline exposure on initiation of EHT, we generated abundant fully complemented or uncomplemented human FA HPCs. These HPCs recapitulated key hallmarks of FA.28,35,37 We identified GAS6 as a novel target of hyperactive p53 activity in FA HPCs and demonstrated that modulation of GAS6 signaling improves the function of FANCA-deficient HPCs.
Our system overcomes challenges faced over the past decade in the development of an IPSC-based human model of FA. Previous systems of hematopoietic differentiation have used either modified reprogramming methods or constitutive complementation of FA patient cells to generate IPSCs.18-20,50 Alternatively, RNA interference has been used in non-FA pluripotent stem cells to disrupt the FA pathway.51 Reprogramming under hypoxic conditions and with the addition of further transcription factors over the minimum required factors has been reported to enhance the efficiency of IPSC derivation from FA patient fibroblasts, although IPSCs from only 2 of 6 patient-derived fibroblast cultures could be propagated in this study.20 Using this approach, post-reprogramming complementation was used to generate independent parallel control cell lines for use in functional analysis.20 Although these approaches yield IPSCs that can undergo hematopoietic differentiation,18 these cells maintain complementation constitutively, and so hematopoietic phenotypes caused by deficiency of the FA pathway are not evaluable and mechanisms of HPC dysfunction cannot be pursued. Alternatively, in rare uncomplemented FA patient-derived IPSC lines, the FA pathway can be repaired after reprogramming in order to generate isogenic, FA-expressing cells.52 However, using our approach, one can readily derive isogenic FA-deficient and FA-competent hematopoietic cells from a single IPSC source. This system modulates FANCA expression at the time of hematopoietic specification and HPC emergence, mitigating confounding effects of FANCA deficiency on cells in the pluripotent state or during early germ layer patterning in EBs. Perhaps most importantly, this approach avoids comparison of IPSC lines derived from FA patients to those from healthy donors, where background genetic variability is well known to affect comparison of differentiation capacity.24,53
Our IPSC-derived, FANCA-deficient HPCs recapitulate key aspects of hallmark FA hematopoietic phenotypes. We found a highly reproducible defect in hematopoietic colony formation in HPCs derived from EBs and the StemDiff 2-dimensional differentiation system, as has been reported with primary patient FA cells.28 Moreover, we find that FANCA-deficient, IPSC-derived HPCs show impaired cell cycle progression as has been demonstrated in both mouse models of FA and primary human cells,54-56 as well as an impaired response to genotoxic stress.57 Although the classic cell cycle defect in FA cells is a G2/M arrest, our findings of slowed G1/S progression are consistent with prior observations of cell cycle defects in FA HPCs.54,58 These results indicate that FA pathway deficiency may exert context-dependent effects on the cell cycle, in this case enhancing the G1/S arrest that occurs during the transition from cycling multipotent/oligopotent progenitor to committed erythroid or myeloid progenitor.44
We used RNA sequencing to delineate the transcriptional divergence of otherwise isogenic FANCA-deficient and FANCA-expressing HPCs. Consistent with the phenotype of impaired clonogenicity, we found that FANCA-expressing HPCs maintained a signature of normal blood stem and progenitor cells relative to FANCA-deficient HPCs. Furthermore, distinct signatures related to cell cycle progression and DNA repair reflected known biology of the FA pathway.54 Using unbiased GSEA against the Molecular Signatures Database,30 we observed that FANCA-deficient HPCs showed a signature of erythroid differentiation relative to FANCA-expressing HPCs, leading us to validate this finding in functional studies. FANCA-deficient HPCs showed accelerated erythroid differentiation compared with FA-competent HPCs, a phenotype not previously described in FA. However, mice with germline FA pathway defects show depletion of phenotypic HPC populations with growth arrest that could represent rapid differentiation toward committed downstream lineages.4,56 The combination of impaired colony-forming capacity and accelerated differentiation in FANCA-deficient HPCs supports a model wherein these cells rapidly lose HPC identity and more readily acquire a lineage-committed state. These findings illustrate the advantage of this system in providing a renewable source of human FA HPCs and isogenic control cells, as such primary patient derived HPCs are difficult to reproducibly investigate disease mechanisms.
FANCA-deficient HPCs show a delayed G1/S phase transition with a diminished proportion of cells in S-phase compared with controls, recapitulating such findings observed in primary human FA CD34+ HPCs.58 As reported previously, we find that this effect is associated with elevated activity of the p53-p21 axis, consistent with recapitulation of human FA disease phenotypes.58 Intact MDM2-mediated p53 inhibition preserved HPC clonogenicity in FANCA-expressing cells, consistent with prior reports demonstrating that p53 activation in FA pathway deficiency impairs the growth and survival of HPCs.59,60 In contrast, activation of p53 by pharmacologically impairing its interaction with MDM2 led to the loss of clonogenicity and acceleration of erythroid differentiation in FANCA-expressing HPCs. Conversely, disruption of the TP53 gene in FANCA-deficient cells rescues clonogenicity and abates terminal differentiation. Given the known requirement of p21 activation for early erythroid commitment,45 we suggest that the attenuated G1/S transition induced by FANCA deficiency predisposes these cells to differentiation. Modulation of p53/p21 activity downstream of FA pathway mutations as a therapeutic approach should be considered with caution, given the accelerated tumorigenesis of p53-null mice bearing FA pathway mutations.61 However, our results concur with the broader paradigm that cell cycle progression regulates differentiation propensity of stem and progenitor cells in a variety of tissues.62,63
We aimed to leverage our model of human FA to identify novel mechanisms of HPC dysregulation that could be targeted therapeutically. We found that hyperactive p53 in FANCA-deficient HPCs induced activation of GAS6, likely by directly binding the GAS6 locus. Since GAS6 acts as a prosurvival, prohematopoietic factor,48,49 we treated FANCA-deficient HPCs with recombinant GAS6, finding that this intervention was sufficient to partially rescue clonogenesis. These results are consistent with leveraging an endogenous negative feedback mechanism wherein potentiation of p53-induced GAS6 signaling may provide an intervention to improve hematopoiesis in FA, although this would in theory have to be accomplished in a selective manner given GAS6’s reported protumorigenic effects.64
Compared with other diseases modeled with patient-derived IPSCs, FA has proven challenging because of the requirement for an intact FA DNA repair pathway for efficient reprogramming of somatic cells to pluripotency.65 We used a doxycycline inducible system for inducible, reversible complementation of FANCA-deficient patient derived human IPSCs. This approach allowed us to generate isogenic FANCA-deficient and FANCA-expressing human HPCs that recapitulated key aspects of FA pathobiology, providing an indefinitely renewable source of human FA and otherwise isogenic control HPCs not obtainable in prior human IPSC-based models of FA. We used this system to discover a novel phenotype wherein FANCA-deficient, IPSC-derived HPCs undergo accelerated erythroid differentiation via a mechanism involving cell cycle regulation by p53 and p21. Our model provides a foundation for future studies of IPSC-based cellular therapy that could benefit patients with FA.
For data sharing requests, e-mail corresponding author, R. Grant Rowe (grant_rowe@dfci.harvard.edu).
Acknowledgments
This study was supported by National Institutes of Health, National Institute of Diabetes, Digestive, and Kidney Diseases grants 1 K08 DK114527-01 (R.G.R.) and U54DK110805-02 (G.Q.D. and T.M.S.), and the Fanconi Anemia Research Fund (R.G.R. and G.Q.D.). S.I.W. was supported by National Institutes of Health, National Cancer Institute grant R01 CA102357 and a grant from the Fanconi Anemia Research Fund. E.L.d.R. was supported by a fellowship from the Coordination for the Improvement of Higher Education, Brazil.
Authorship
Contribution: W.M., S.B., S.R.-T., V.M., V.L., S.C., A.M.Z., O.A., C.K., Y.Z., T.M.S., and R.G.R. performed experiments; B.L.E., A.S., S.R.-T. and S.I.W. provided key reagents. S.B., S.R.-T., S.I.W., E.L.d.R., T.M.S., and R.G.R. analyzed data; and T.E.N., G.Q.D., S.I.W., and R.G.R. designed the research and wrote the manuscript.
Conflict-of-interest disclosure: During the conduct of this study, G.Q.D. held equity or received consulting fees from the following: Epizyme, 28/7 Therapeutics, and MPM Capital, LLC. The remaining authors declare no competing financial interests.
The current affiliation for E.L.d.R. is Department of Microbiology, Immunology, and Parasitology, Federal University of Santa Catarina, Florianopolis, Brazil.
The current affiliation for V.L. is Center for Hematology and Regenerative Medicine, Karolinska Institutet, Department of Medicine, Karolinska University Hospital, Huddinge, Stockholm, Sweden.
Correspondence: R. Grant Rowe, Karp Family Research Building, 7th Floor, 1 Blackfan Cir, Boston, MA 02115; e-mail: grant_rowe@dfci.harvard.edu.

