

Key Points
RNA-seq can be implemented into a clinical service to inform risk stratification of patients with nonstandard molecular features.
RNA-seq data can be used to identify IKZF1 deletions.

Abstract
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy, and implementation of risk-adapted therapy has been instrumental in the dramatic improvements in clinical outcomes. A key to risk-adapted therapies includes the identification of genomic features of individual tumors, including chromosome number (for hyper- and hypodiploidy) and gene fusions, notably ETV6-RUNX1, TCF3-PBX1, and BCR-ABL1 in B-cell ALL (B-ALL). RNA-sequencing (RNA-seq) of large ALL cohorts has expanded the number of recurrent gene fusions recognized as drivers in ALL, and identification of these new entities will contribute to refining ALL risk stratification. We used RNA-seq on 126 ALL patients from our clinical service to test the utility of including RNA-seq in standard-of-care diagnostic pipelines to detect gene rearrangements and IKZF1 deletions. RNA-seq identified 86% of rearrangements detected by standard-of-care diagnostics. KMT2A (MLL) rearrangements, although usually identified, were the most commonly missed by RNA-seq as a result of low expression. RNA-seq identified rearrangements that were not detected by standard-of-care testing in 9 patients. These were found in patients who were not classifiable using standard molecular assessment. We developed an approach to detect the most common IKZF1 deletion from RNA-seq data and validated this using an RQ-PCR assay. We applied an expression classifier to identify Philadelphia chromosome–like B-ALL patients. T-ALL proved a rich source of novel gene fusions, which have clinical implications or provide insights into disease biology. Our experience shows that RNA-seq can be implemented within an individual clinical service to enhance the current molecular diagnostic risk classification of ALL.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignancy of childhood, accounting for 26% of pediatric cancers.1 Survival rates for children diagnosed with ALL have dramatically improved to >90%, as a result of modern chemotherapy, risk-adapted therapeutic regimens, and the development of molecularly targeted therapies.2 B-cell ALL (B-ALL) comprises 80% of pediatric ALL and, in recent years, has been extensively characterized in large cohort studies using next-generation sequencing methodologies.3-7 This has greatly expanded the number of recurrent driver genomic lesions recognized in B-ALL. Although not all of these lesions are currently incorporated into the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia, it is likely that, over time, many will contribute to risk stratification and therapy selection.8,9 This presents clinical services with the challenge of identifying new B-ALL entities in a timely and accurate manner. Specific molecular risk factors have yet to be incorporated into treatment stratification of T-cell ALL (T-ALL). However, as sequencing identifies new molecular entities of T-ALL and early T-cell precursor ALL (ETP-ALL), the possibility of treatment stratification based on genomic findings emerges, which may reduce the increased rates of treatment failure and relapse associated with T-ALL.10,11
The clinical features of “high-risk” B-ALL include age at diagnosis (≥10 years) and/or disease burden indicated by white blood cell (WBC) count ≥50 × 109/L.12 Additional risk stratification of B-ALL patients after induction chemotherapy incorporates cytogenetic features and early response to therapy, minimal residual disease (MRD). These are now standard of care and have contributed to improving ALL outcomes.13-15 Low-risk features of ALL include ETV6-RUNX1 and favorable chromosomal trisomies (chromosomes 4 and 10 in particular), whereas very high-risk features include hypodiploidy (<44 chromosomes), BCR-ABL1, TCF3-HLF, and positive MRD after induction.13,16,17 MRD-directed treatment intensification has improved outcomes in patients with high-risk cytogenetic features.7,18
RNA-sequencing (RNA-seq) has been instrumental in establishing the landscape of driver gene fusions in pediatric ALL.7,19-21 Philadelphia chromosome (Ph)–like ALL is a prescient example of the importance of new ALL entities and the clinical impact of early recognition. It was included in the 2016 WHO guidelines as a provisional B-lymphoblastic leukemia/lymphoma entity, defined by gene expression profile, and is commonly associated with the presence of chromosomal rearrangements involving cytokine receptor or tyrosine kinase genes.8 This ALL subtype is potentially targetable with tyrosine kinase inhibitors. Other structural variants, notably IKZF1 deletions, also contribute to the estimated risk of treatment failure or relapse.5,6,22-24
In this study we performed RNA-seq on a clinical ALL cohort, in parallel with standard-of-care testing, to establish how reliably oncogenic driver fusions, Ph-like expression profiles and structural variants, such as IKZF1 deletions, can be detected. Our experience suggests RNA-seq will have the greatest clinical impact when applied to patients in whom standard molecular testing is negative. In this group, RNA-seq will identify previously unsuspected driver fusions. RNA-seq data can also be used to identify common IKZF1 deletions. This requires specific analysis of IKZF1 isoforms and cannot be deduced from simple IKZF1 expression levels.
Materials and methods
Study design
This study was approved by the Royal Children’s Hospital Human Research Ethics Committee (HREC 34127). We retrospectively sequenced 126 patients who were diagnosed with ALL from September 2009 through August 2018. The median follow-up time was 2.57 years, reflecting the recent diagnosis of most of our patient cohort. We initially sequenced all patients diagnosed with ALL at the beginning of the trial but moved to select only patients that were negative in standard-of-care cytogenetics (nonstandard) for sequencing. Patients were classified as “defined” if they fit 1 of the 7 established subtypes of B-lymphoblastic leukemia/lymphoma or the provisional entity ETP-ALL, as described in the WHO 2016 revision of the classification of myeloid neoplasms and acute leukemias.8 Follow-up measures included death, relapse, and bone marrow transplantation. Length of follow-up varied, given the recent diagnosis for most of our patient cohort. Data were locked on 18 December 2018.
Standard-of-care diagnostics
All patient samples had been analyzed by G-banded karyotyping and targeted fluorescence in situ hybridization (FISH) performed at Victorian Clinical Genetics Services, and DNA index was determined by flow cytometry as part of standard-of-care clinical diagnostics. Methodological details are included in the supplemental Materials and methods.
Nonstandard molecular analysis
A single-nucleotide polymorphism (SNP) microarray was performed at Victorian Clinical Genetics Services, and real-time quantitative polymerase chain reaction (RQ-PCR) was performed at Children’s Cancer Institute (supplemental Materials and methods).
RNA-seq
Patient blood and bone marrow samples were obtained from the Children’s Cancer Centre Tissue Bank, Murdoch Children’s Research Institute. The details of RNA extraction, library preparation, and sequencing parameters are provided in the supplemental Materials and methods.
Fusion detection
Detection of the fusion genes was performed using JAFFA v1.08 in the direct mode.25 JAFFA uses unaligned fastq.gz files as the input data. Details on filtering of fusion transcripts identified by JAFFA are provided in supplemental Materials and methods. Visualization of fusion genes was performed using Clinker v1.32 and v1.4,26 to graphically depict sequencing coverage, genomic breakpoints, and transcript variants. A second fusion-detection program, Arriba v1.1.0 (https://github.com/suhrig/arriba/), was run on the RNA-seq for cases where a known fusion was missed by JAFFA. See supplemental Materials and methods for details.
Gene expression analysis
RNA sequence fastq.gz files were aligned to the human genome (hg19) using the STAR aligner (version 2.5.2a) with default parameters in the 2-pass mode, with the exception of parameters controlling junctions limitOutSJcollapsed and limitSjdbInsertNsj set to 5 000 000 and 2 000 000, respectively, and chimeric alignment parameters of chimSegmentMin and chimSegmentReadGapMax set to 10 and 3, respectively. featureCounts (v1.50) was used to summarize the reads across the genes, counting only uniquely mapped genes by using a parsed version of the comprehensive GENCODE 19 annotation.27 For individual gene expression analysis read counts were normalized by library size and log2 transformed.
Detecting IKZF1 deletions using RNA-seq data
The full details are provided in supplemental Materials and methods. In brief, a custom reference transcriptome with additional transcripts corresponding to the deletions of IKZF1 exons 4-7, 2-7, 2-8, and 4-8 was added to the ENSEMBL release 96 human transcriptome reference and their expression level estimated with Salmon v0.12.0.28 We then estimated the abundance (transcripts per million) of each transcript and of all IKZF1 transcripts. Scripts to identify IKZF1 deletions are available at https://github.com/Oshlack/ALL-RNAseq-utility-paper.
Gene expression classifier
Classification based on gene expression profiles for each sample was performed using the AllSorts classifier for B-ALL (https://github.com/Oshlack/AllSorts). AllSorts consists of a random forest trained on 484 sets of B-ALL RNA-seq data, with 289 samples provided from Roberts et al7 and 195 samples from Lilljebjörn et al24 (European Genome-phenome Archive: EGAD00001002112). Details are included in supplemental Materials and methods.
Results
Clinical characteristics and cytogenetic features of the ALL cohort
The ALL cohort consisted of children treated at the Royal Children’s Hospital, with biobanked samples in the Children’s Cancer Centre Tissue Bank and informed consent for sequencing analysis. All cases had standard-of-care molecular diagnostic analyses including karyotype; FISH analysis for ETV6-RUNX1, BCR-ABL1, and KMT2A rearrangements; and DNA ploidy by flow cytometry. Our 126 samples represented a selected subset deliberately biased toward samples without a positive standard-of-care molecular diagnostic classification. The clinical and cytogenetic characteristics of the cohort are shown in Table 1 and are broadly consistent with established features of childhood ALL.29,30 All B-ALL cases were B-cell precursor ALL, and 4 of 27 (14.8%) T-ALL cases were ETP-ALL. National Cancer Institute (NCI) risk was calculated for the B-ALL patients. In total, our cohort comprised 62.6% standard-risk and 37.4% high-risk B-ALL patients.
Patient clinical characteristics
| Patients | ||||
|---|---|---|---|---|
| B-ALL (n = 99) | T-ALL (n = 27) | |||
| n | % | n | % | |
| Sex | ||||
| Male | 43 | 43.4 | 21 | 77.8 |
| Female | 56 | 56.6 | 6 | 22.2 |
| Age, y | ||||
| <1 | 4 | 4.0 | 0 | 0.0 |
| 1-9.99 | 77 | 77.8 | 19 | 70.4 |
| ≥10 | 18 | 18.2 | 8 | 29.6 |
| WBC, ×109/L | ||||
| <50 | 84 | 84.8 | 11 | 40.7 |
| ≥50 | 15 | 15.2 | 16 | 59.3 |
| NCI risk | ||||
| Standard | 62 | 62.6 | — | — |
| High | 37 | 37.4 | — | — |
| Extramedullary disease | 12 | 12.1 | 3 | 11.1 |
| Trisomy 21 | 5 | 5.1 | 0 | 0.0 |
| WHO subtype classification* | ||||
| Defined | ||||
| Hyperdiploid | 29 | 29.3 | — | — |
| Hypodiploid | 5 | 5.0 | — | — |
| BCR-ABL1 | 4 | 4.0 | — | — |
| KMT2A-rearranged | 6 | 6.1 | — | — |
| ETV6-RUNX1 | 20 | 20.2 | — | — |
| TCF3-PBX1 | 2 | 2.0 | — | — |
| IL3-IGH | 1 | 1.0 | — | — |
| Provisional entities | ||||
| Early T-cell precursor ALL | — | — | 4 | 14.8 |
| Undefined | 32 | 32.3 | 23 | 85.2 |
| MRD status at the end of induction† | ||||
| Positive | 35 | 35.4 | 18 | 66.7 |
| Negative | 64 | 64.6 | 9 | 33.3 |
| MRD status at the end of consolidation† | ||||
| Positive | 8 | 8.1 | 9 | 33.3 |
| Negative | 91 | 91.9 | 18 | 66.7 |
| Bone marrow transplantation | ||||
| Total number of patients | 11 | 11.1 | 6 | 22.2 |
| CR1 | 3 | 3.0 | 6 | 22.2 |
| CR2 | 8 | 8.1 | 1 | 3.7 |
| Outcome | ||||
| Death | 9 | 9.1 | 0 | 0.0 |
| Relapse | 16 | 16.2 | 1 | 3.7 |
| No event | 79 | 79.8 | 26 | 96.3 |
| Patients | ||||
|---|---|---|---|---|
| B-ALL (n = 99) | T-ALL (n = 27) | |||
| n | % | n | % | |
| Sex | ||||
| Male | 43 | 43.4 | 21 | 77.8 |
| Female | 56 | 56.6 | 6 | 22.2 |
| Age, y | ||||
| <1 | 4 | 4.0 | 0 | 0.0 |
| 1-9.99 | 77 | 77.8 | 19 | 70.4 |
| ≥10 | 18 | 18.2 | 8 | 29.6 |
| WBC, ×109/L | ||||
| <50 | 84 | 84.8 | 11 | 40.7 |
| ≥50 | 15 | 15.2 | 16 | 59.3 |
| NCI risk | ||||
| Standard | 62 | 62.6 | — | — |
| High | 37 | 37.4 | — | — |
| Extramedullary disease | 12 | 12.1 | 3 | 11.1 |
| Trisomy 21 | 5 | 5.1 | 0 | 0.0 |
| WHO subtype classification* | ||||
| Defined | ||||
| Hyperdiploid | 29 | 29.3 | — | — |
| Hypodiploid | 5 | 5.0 | — | — |
| BCR-ABL1 | 4 | 4.0 | — | — |
| KMT2A-rearranged | 6 | 6.1 | — | — |
| ETV6-RUNX1 | 20 | 20.2 | — | — |
| TCF3-PBX1 | 2 | 2.0 | — | — |
| IL3-IGH | 1 | 1.0 | — | — |
| Provisional entities | ||||
| Early T-cell precursor ALL | — | — | 4 | 14.8 |
| Undefined | 32 | 32.3 | 23 | 85.2 |
| MRD status at the end of induction† | ||||
| Positive | 35 | 35.4 | 18 | 66.7 |
| Negative | 64 | 64.6 | 9 | 33.3 |
| MRD status at the end of consolidation† | ||||
| Positive | 8 | 8.1 | 9 | 33.3 |
| Negative | 91 | 91.9 | 18 | 66.7 |
| Bone marrow transplantation | ||||
| Total number of patients | 11 | 11.1 | 6 | 22.2 |
| CR1 | 3 | 3.0 | 6 | 22.2 |
| CR2 | 8 | 8.1 | 1 | 3.7 |
| Outcome | ||||
| Death | 9 | 9.1 | 0 | 0.0 |
| Relapse | 16 | 16.2 | 1 | 3.7 |
| No event | 79 | 79.8 | 26 | 96.3 |
CR1/CR2, complete remission-1 and -2.
WHO subtype classification based on karyotype and FISH analysis performed as part of standard clinical diagnostic testing.
MRD positive defined as ≥0.01% leukemic cells measured by flow cytometry. Where flow cytometry was uninformative, quantitative real-time PCR and molecular markers were used to determine MRD status.
MRD was assessed at the end of induction (EOI; day 29) and the end of consolidation (EOC), by flow cytometry or molecular methods. The MRD status of our cohort is shown in Table 1. The rates of positive MRD at EOI and EOC are higher when compared with all ALL patients managed in our service reflecting selection of nonstandard cases. Nine patients (7.1%) died during the follow-up period, and the median follow-up time for the remaining patients was 2.57 years.
Fusion genes identified in B-ALL
The RNA-seq results in B-ALL are summarized in Figure 1. After standard molecular subclassification, 67 samples conformed to a defined subtype of B-lymphoblastic leukemia/lymphoma. Additional genomic lesions were identified in only 3 of those samples by RNA-seq: 2 PAX5 out-of-frame fusions indicative of PAX5 deletion and the specific characterization of an uncommon KMT2A-USP2 fusion. This KMT2A fusion is an example of an emerging class of rearrangements with an alternative KMT2A breakpoint.31 Of the 32 “undefined” samples after standard molecular testing, 17 had karyotypic or FISH findings suggestive of provisional or emerging B-ALL subtypes. In all but 1 instance, RNA-seq data definitively demonstrated the presence of these structural variants. They included 2 rare ABL1 fusions, 8 P2RY8-CRLF2 rearrangements, 4 nonstandard ETV6 rearrangements, 1 TCF3-ZNF384 rearrangement, and 1 PAX5-AUTS2 rearrangement.32 The 1 missed structural variant was an IGH-CRLF2 variant. Fusions involving nontranscribed regions of the genome, such as the IGH promotor or enhancer elements, are difficult to detect with RNA-seq.
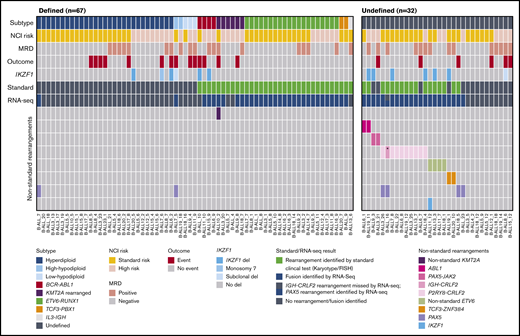
Fusion genes identified in B-ALL patients. Data are presented for 99 patients with B-ALL on whom we performed RNA-seq. Each column represents a single patient. Patients were classified as “Defined” if they fell into a WHO-established subtype (n = 67) or “Undefined” (n = 32), according to the key. NCI risk classification, MRD at the end of induction, outcome data, where event indicates death or relapse, and IKZF1 status, determined by microarray or RQ-PCR assay, are shown for each patient and colored according to the key. Rearrangements are identified by standard clinical test (standard) including G-banding analysis (karyotype) or FISH analysis and are shown in green. The RNA-seq row denotes fusions identified by RNA-seq using JAFFA and are shown in blue. The gray and blue bar represents a sample in which an IGH-CRLF2 rearrangement, identified by karyotype, was missed by RNA-seq but a PAX5 rearrangement was identified by RNA-seq. Nonstandard rearrangements identified are colored according to the key. *IGH-CRLF2 rearrangement identified by karyotype and not identified by RNA-seq.
Fusion genes identified in B-ALL patients. Data are presented for 99 patients with B-ALL on whom we performed RNA-seq. Each column represents a single patient. Patients were classified as “Defined” if they fell into a WHO-established subtype (n = 67) or “Undefined” (n = 32), according to the key. NCI risk classification, MRD at the end of induction, outcome data, where event indicates death or relapse, and IKZF1 status, determined by microarray or RQ-PCR assay, are shown for each patient and colored according to the key. Rearrangements are identified by standard clinical test (standard) including G-banding analysis (karyotype) or FISH analysis and are shown in green. The RNA-seq row denotes fusions identified by RNA-seq using JAFFA and are shown in blue. The gray and blue bar represents a sample in which an IGH-CRLF2 rearrangement, identified by karyotype, was missed by RNA-seq but a PAX5 rearrangement was identified by RNA-seq. Nonstandard rearrangements identified are colored according to the key. *IGH-CRLF2 rearrangement identified by karyotype and not identified by RNA-seq.
Fifteen samples were without an identified lesion, either established or suspected, by standard molecular testing (Figure 1). In one-third of these, RNA-seq identified a known or potential driver rearrangement. Strikingly, this included 2 samples expressing PAX5-JAK2 fusions, which are potentially therapeutically targetable.33 RNA-seq also identified individual samples with a P2RY8-CRLF2, TCF3-ZNF384, and PAX5-AUTS2 fusion. With the addition of RNA-seq, 10 samples remained undefined. These data show that in B-ALL, the greatest yield from RNA-seq will be in the subset of patients in whom standard molecular testing does not definitively identify a B-ALL molecular subtype.
RNA-seq also identified novel ETV6 fusion events, which disrupt the coding domains of ETV6 (supplemental Figure 1). One of these included a CHST11-VPS8 fusion transcript, likely the result of a more complex chromosome (Chr)3/Chr12 rearrangement also involving ETV6. We additionally identified PAX5 fusion events in 6 patients, 4 of which were classified by another molecular feature (supplemental Table 3). These fusions likely arise from PAX5 deletion events.
Gene rearrangements and fusion genes identified in T-ALL
We sequenced 27 T-ALL cases (Figure 2A). Four of these were defined as ETP-ALL on immunophenotype, and the remaining 23 were classified as undefined. Conventional karyotype and FISH identified chromosomal rearrangements in 6 samples in the undefined group, including 2 TRA-LMO2 rearrangements; 1 KMT2A-MLLT1, 1 NUP214-ABL1, and 1 MYC rearrangement; and 1 TRB-TLX1 rearrangement. The TRB-TLX1 case also harbored a deletion on Chr10 that resulted in the expression of a novel PTEN-ATAD fusion transcript detected by RNA-seq.
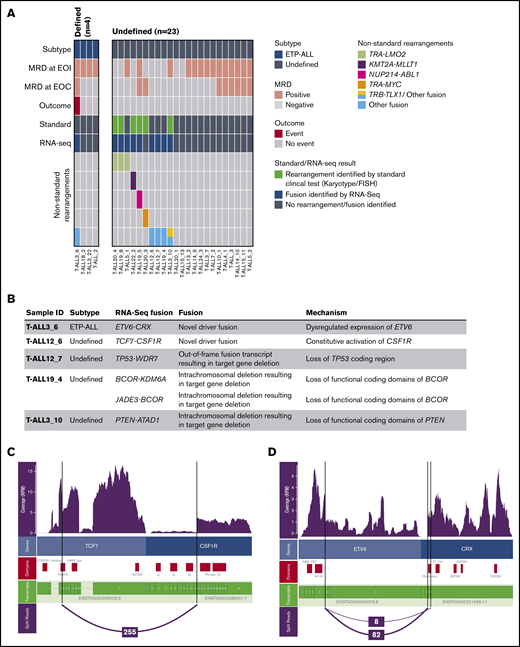
Gene rearrangements and fusion genes identified in T-ALL patients. (A) Data are presented for 27 patients with T-ALL on whom we performed RNA-seq. Each column represents a single patient. Patients who were classified into the WHO provisional subtype early T-cell precursor ALL (ETP-ALL) are classified as defined (n = 4), and the remaining patients were classified as undefined (n = 23). MRD at EOI and EOC, and outcome data, where event indicates death or relapse, are shown for each patient and colored according to the key. Rearrangements identified by standard clinical test (standard) include gene rearrangements identified by either karyotype or FISH
Gene rearrangements and fusion genes identified in T-ALL patients. (A) Data are presented for 27 patients with T-ALL on whom we performed RNA-seq. Each column represents a single patient. Patients who were classified into the WHO provisional subtype early T-cell precursor ALL (ETP-ALL) are classified as defined (n = 4), and the remaining patients were classified as undefined (n = 23). MRD at EOI and EOC, and outcome data, where event indicates death or relapse, are shown for each patient and colored according to the key. Rearrangements identified by standard clinical test (standard) include gene rearrangements identified by either karyotype or FISH
Seventeen patients had no rearrangement identified by standard of care. RNA-seq identified a TRA-LMO2 rearrangement in 1 patient and range of novel structural variants that included candidate driver fusions or disruptions of tumor suppressors (Figure 2B). The tumor-suppressor disruptions included an expressed TP53-WDR7 fusion that juxtaposes the 5′ untranslated region of TP53 with WDR7, predicted to result in the loss of functional TP53, and the previously described PTEN-ATAD fusion transcript (supplemental Figure 2). Two BCOR fusions identified in the same sample (Figure 2B) suggested a complex structural event involving ChrX. BCOR loss-of-function variants are a recognized feature in T-ALL.34
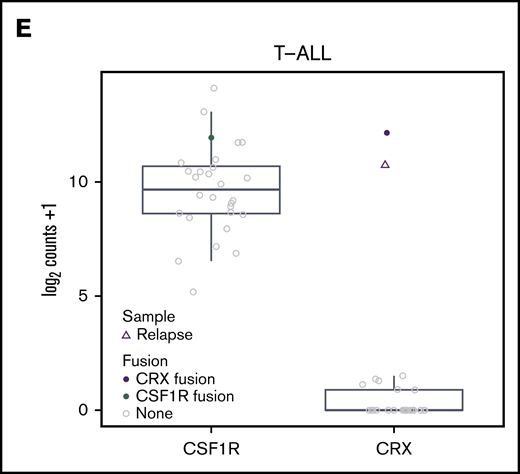
Two novel candidate T-ALL driver fusions were detected by RNA-seq: TCF7-CSF1R (Figure 2C) and ETV6-CRX (Figure 2D). Expression of the cone–rod homeobox (CRX) gene is normally restricted to photoreceptor cells specifically, and the in-frame fusion we identified drives expression of the CRX homeobox domain. This sample had the highest expression of CRX of any sample sequenced (Figure 2E). Further, the fusion was highly expressed in both the diagnostic and relapse sample (Figure 2E). We speculate that enforced expression of this homeobox domain blocks differentiation in the ETP-ALL. The TCF7-CSF1R fusion transcript contains the entire tyrosine kinase–coding domain of CSF1R, driving increased expression of CSF1R (Figure 2C,E). Although this patient responded well to standard chemotherapy, a future relapse may be amenable to dasatinib treatment.33,35 We observed high CSF1R expression in 4 additional samples, but did not identify any rearrangements involving CSF1R using an alternative method of detection, Arriba36 (supplemental Table 4). Together, these data show that RNA-seq has a relatively high yield of identifying novel and potentially targetable driver fusions in pediatric T-ALL.
Elevated gene expression is indicative of some, but not all, fusion transcripts
Elevated expression of driver genes encoded by gene fusions may provide orthogonal evidence of rearrangements that are difficult to detect or are missed by RNA-seq. Fusion transcripts were identified in 48 of 56 (85.7%) patient samples in which the same rearrangement had been identified or suspected using standard-of-care cytogenetics (Table 2). However, 8 standard rearrangements were missed by RNA-seq. The most consequential of these was a BCR-ABL1 fusion in a known Ph+ B-ALL (B-ALL_10), most likely the result of low leukemic blast content (6%) in the sample that was taken 8 days after commencement of induction chemotherapy. The alternative fusion finding algorithm, Arriba, detected a single BCR-ABL1 spanning read. We explored this further by diluting RNA from a BCR-ABL1 positive B-ALL sample into RNA extracted from a healthy control and sequencing at each dilution. When the BCR-ABL1 positive RNA represented 10% or less of the total sample, the ability to detect BCR-ABL fusion was lost (supplemental Figure 3). In addition, 2 IGH rearrangements, IGH-CRLF2 and IL3-IGH, were identified by karyotype but not by RNA-seq, using either JAFFA or Arriba (Table 2; supplemental Table 3). We anticipated that IGH rearrangements would be challenging to detect by RNA-seq, as these rearrangements result in target gene expression driven by an upstream IGH enhancer element, but do not result in the expression of abnormal fusion transcripts.
Comparison of RNA-seq to standard-of-care cytogenetic testing to identify fusion genes
| Rearrangements identified | Standard test, n | RNA-seq (additional findings to standard testing), n | RNA-seq/standard test, % | Comment | |
|---|---|---|---|---|---|
| B-ALL | |||||
| BCR-ABL1 | 4 | 3 | 75 | Missed BCR-ABL1 (1). RNA-Seq performed on sample taken 8 d after commencement of induction. | |
| KMT2A rearrangement | 6 | 4 | 66.7 | Missed KMT2A-MLLT1 (1) and KMT2A-X (1). Low expression of KMT2A in these samples (Figure 3A) | |
| ETV6-RUNX1 | 20 | 20 | 100 | — | |
| TCF3-PBX1 | 2 | 2 | 100 | — | |
| IL3-IGH | 1 | 0 | 0 | Missed IL3-IGH (1) | |
| Undefined/B-other | |||||
| ABL1 rearrangement | 2 | 2 | 100 | — | |
| JAK2 rearrangement | 0 | 0 (2) | — | PAX5-JAK2 (2) | |
| CRLF2 rearrangement | 9 | 8 (1) | 88.9 | Missed IGH-CRLF2 (1). P2RY8-CRLF2 (1) | |
| ETV6 rearrangement | 4 | 3 (2) | 75 | Missed ETV6 rearrangement (1). CHST11-VSP8 (1)fusion found in this patient by RNA-Seq. Suggesting of a more complex Chr3/12 rearrangement. Two ETV6 rearrangements found in 1 patient: ETV6-MDH2 + ETV6-ATP5SL (1) | |
| ZNF384 rearrangement | 1 | 1 (1) | 100 | TCF3-ZNF384 (1) | |
| PAX5 rearrangement | 1 | 1 (1) | 100 | PAX5-AUTS2 (1) | |
| Additional findings in patients otherwise classified | 0 | 0 (5) | — | PAX5-LINC01400 (hyperdiploid, 1); PAX5-ARHGAP22 (hypodiploid, 1); PAX5-NOL4L (P2RY8-CRLF2, 1); PAX5-MSMP (IGH-CRLF2, 1); IKZF1-CEP170+ADSS-IKZF1 (ETV6-CLN6, 1) | |
| T-ALL | |||||
| Undefined/T-other | |||||
| LMO2 rearrangement | 2 | 2 (1) | 100 | LMO2-TRA (1) | |
| KMT2A rearrangement | 1 | 1 | 100 | — | |
| NUP214-ABL1 | 1 | 1 | 100 | — | |
| MYC rearrangement | 1 | 0 | 0 | Missed TRA-MYC (1) | |
| Other | 1 | 0 (4) | 0 | PTEN-ATAD1 (1); BCOR-KDM6A+PHF16-BCOR (1); TCF7-CSF1R (1); TP53-WDR7 (1) | |
| Additional findings in patients otherwise classified | 0 | 0 (1) | — | ETV6-CRX (ETP-ALL,1) | |
| Total | 56 | 48 (18) | 85.7 | ||
| Rearrangements identified | Standard test, n | RNA-seq (additional findings to standard testing), n | RNA-seq/standard test, % | Comment | |
|---|---|---|---|---|---|
| B-ALL | |||||
| BCR-ABL1 | 4 | 3 | 75 | Missed BCR-ABL1 (1). RNA-Seq performed on sample taken 8 d after commencement of induction. | |
| KMT2A rearrangement | 6 | 4 | 66.7 | Missed KMT2A-MLLT1 (1) and KMT2A-X (1). Low expression of KMT2A in these samples (Figure 3A) | |
| ETV6-RUNX1 | 20 | 20 | 100 | — | |
| TCF3-PBX1 | 2 | 2 | 100 | — | |
| IL3-IGH | 1 | 0 | 0 | Missed IL3-IGH (1) | |
| Undefined/B-other | |||||
| ABL1 rearrangement | 2 | 2 | 100 | — | |
| JAK2 rearrangement | 0 | 0 (2) | — | PAX5-JAK2 (2) | |
| CRLF2 rearrangement | 9 | 8 (1) | 88.9 | Missed IGH-CRLF2 (1). P2RY8-CRLF2 (1) | |
| ETV6 rearrangement | 4 | 3 (2) | 75 | Missed ETV6 rearrangement (1). CHST11-VSP8 (1)fusion found in this patient by RNA-Seq. Suggesting of a more complex Chr3/12 rearrangement. Two ETV6 rearrangements found in 1 patient: ETV6-MDH2 + ETV6-ATP5SL (1) | |
| ZNF384 rearrangement | 1 | 1 (1) | 100 | TCF3-ZNF384 (1) | |
| PAX5 rearrangement | 1 | 1 (1) | 100 | PAX5-AUTS2 (1) | |
| Additional findings in patients otherwise classified | 0 | 0 (5) | — | PAX5-LINC01400 (hyperdiploid, 1); PAX5-ARHGAP22 (hypodiploid, 1); PAX5-NOL4L (P2RY8-CRLF2, 1); PAX5-MSMP (IGH-CRLF2, 1); IKZF1-CEP170+ADSS-IKZF1 (ETV6-CLN6, 1) | |
| T-ALL | |||||
| Undefined/T-other | |||||
| LMO2 rearrangement | 2 | 2 (1) | 100 | LMO2-TRA (1) | |
| KMT2A rearrangement | 1 | 1 | 100 | — | |
| NUP214-ABL1 | 1 | 1 | 100 | — | |
| MYC rearrangement | 1 | 0 | 0 | Missed TRA-MYC (1) | |
| Other | 1 | 0 (4) | 0 | PTEN-ATAD1 (1); BCOR-KDM6A+PHF16-BCOR (1); TCF7-CSF1R (1); TP53-WDR7 (1) | |
| Additional findings in patients otherwise classified | 0 | 0 (1) | — | ETV6-CRX (ETP-ALL,1) | |
| Total | 56 | 48 (18) | 85.7 | ||
Bold typeface indicates fusions that were identified by RNA-seq only.
We explored the relationship between missed B-ALL and T-ALL fusions and expression of target genes involved in those rearrangements (Figure 3). The highest expressors of IL3 and CRLF2 were those harboring the respective IGH fusions (Figure 3A). Moreover, other CRLF2 fusions (P2RY8-CRLF2) were the next highest cluster of expressors. Only 1 P2RY8-CRLF2 fusion sample had expression at or below the cohort median (B-ALL3_25). A SNP microarray suggested that the PAR1 deletion was subclonal (20%) in this sample. Samples with JAK2 and ABL1 fusions were high expressors of these genes. In T-ALL, the TRB-TLX1 fusion drove TLX1 expression to levels far in excess of the rest of the T-ALL cohort. This was not the case for TRA-MYC (Figure 3B). Arriba identified fusion transcripts evidencing T-cell receptor rearrangements in T-ALL20_3 (TRA-MYC) and T-ALL3_10 (TRB-TLX1; supplemental Table 4).
![Gene expression analysis of genes recurrently altered in B- and T-ALL. (A) Gene expression analysis of all B-ALL samples analyzing expression of ABL1, JAK2, KMT2A, IL3, and CRLF2. Plots show gene names on the x-axis and gene expression (log2 [counts +1]) on the y-axis. Box plots show mean and interquartile range. Samples in which a fusion was identified by RNA-seq involving that particular gene are green. Samples containing rearrangements involving IGH are pink and were missed by RNA-seq (IGHr). All other samples that contain gene fusions missed by RNA-seq are red (Missed). (B) Gene expression analysis of all T-ALL samples showing expression levels of MYC and TLX1. Plots show gene name on the x-axis and gene expression (log2 [counts +1]) on the y-axis. Box plots show mean and interquartile range. Samples in which a fusion was identified by RNA-seq involving MYC or TLX1 are light pink or pink, respectively. All remaining samples are gray.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/5/10.1182_bloodadvances.2019001008/3/m_advancesadv2019001008f3.png?Expires=1769091492&Signature=c2hr0XS4EGanBGPFfcVudSHYmo4cfKz8UQC0IEIjExQziiqdUT~T6Jj0MKM~CyxGXTAAJ0vGViUx7flr26O3AWqvbTq~zaDb2V0p9mlg4MJmmQ9~FdJUGOkvhil1HFxgDwn2cDc0BP3zJSwjg9TK28WIdcV3pZdH5XAz1r2wT5QjpZsXcwU21xbNH-cS2iAqgtdTfv5aEUmAUOFofAe9-fu61JTEo9IjK1hxjUa1ihBojosDV~wSo5UrHrf43QfD6EtebF19Wcj9N20EmX7hX5jXITYxYtMMpKBuXfYYJC3cgvyNTr0FJyH9CDURj-r7A7Y22-rjnO2ya1WHClo3Sg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Gene expression analysis of genes recurrently altered in B- and T-ALL. (A) Gene expression analysis of all B-ALL samples analyzing expression of ABL1, JAK2, KMT2A, IL3, and CRLF2. Plots show gene names on the x-axis and gene expression (log2 [counts +1]) on the y-axis. Box plots show mean and interquartile range. Samples in which a fusion was identified by RNA-seq involving that particular gene are green. Samples containing rearrangements involving IGH are pink and were missed by RNA-seq (IGHr). All other samples that contain gene fusions missed by RNA-seq are red (Missed). (B) Gene expression analysis of all T-ALL samples showing expression levels of MYC and TLX1. Plots show gene name on the x-axis and gene expression (log2 [counts +1]) on the y-axis. Box plots show mean and interquartile range. Samples in which a fusion was identified by RNA-seq involving MYC or TLX1 are light pink or pink, respectively. All remaining samples are gray.
Gene expression analysis of genes recurrently altered in B- and T-ALL. (A) Gene expression analysis of all B-ALL samples analyzing expression of ABL1, JAK2, KMT2A, IL3, and CRLF2. Plots show gene names on the x-axis and gene expression (log2 [counts +1]) on the y-axis. Box plots show mean and interquartile range. Samples in which a fusion was identified by RNA-seq involving that particular gene are green. Samples containing rearrangements involving IGH are pink and were missed by RNA-seq (IGHr). All other samples that contain gene fusions missed by RNA-seq are red (Missed). (B) Gene expression analysis of all T-ALL samples showing expression levels of MYC and TLX1. Plots show gene name on the x-axis and gene expression (log2 [counts +1]) on the y-axis. Box plots show mean and interquartile range. Samples in which a fusion was identified by RNA-seq involving MYC or TLX1 are light pink or pink, respectively. All remaining samples are gray.
Interestingly, KMT2A-rearranged samples tended to have lower expression of KMT2A than samples without KMT2A rearrangements. KMT2A fusions missed in 2 samples by both fusion-detection algorithms were among the lowest expressors of this gene (Figure 3A). We next assessed expression of genes associated with the KMT2A-rearranged transcription program: PBX3, MEF2C, MEIS1, HOXA9, HOXA10, and DOT1L. We showed that HOXA9 and HOXA10 were highly expressed in 4 KMT2A-rearranged samples, including 1 in which the KMT2A rearrangement was not detected by RNA-seq (B-ALL5_7; supplemental Figure 4). This suggests that increased expression of HOXA9 and HOXA10 may indicate the presence of an undetected KMT2A rearrangement. Taken together, these data show that in some instances, gene expression can be a surrogate marker of the presence of an activating fusion. Fusions with low expression, such as KMT2A rearrangements, or low tumor burden will present a challenge to the use of RNA-seq as a single tool to identify ALL fusions.
Common IKZF1 deletions can be reliably detected using RNA-seq
Clinical assays to detect IKZF1 deletions include SNP microarray or RQ-PCR (Figure 4A). IKZF1 deletions were identified clinically in 14 B-ALL (14.1%) and 1 T-ALL patient (3.7%) and are detailed in supplemental Table 5. We investigated how RNA-seq data could be used to identify IKZF1 deletions.
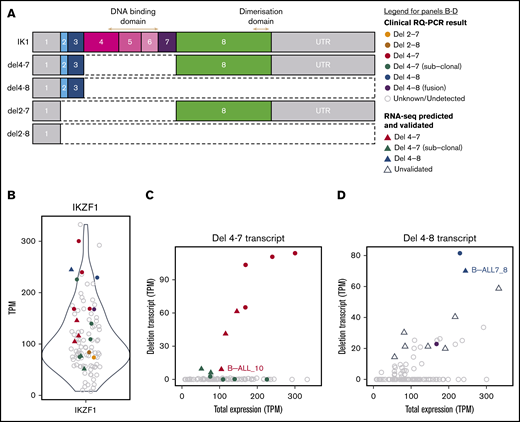
Identifying IKZF1 deletions using RNA-seq data. (A) Schematic of the IKZF1 canonical transcript (IK1) and augmented deletion transcripts, del4-7, del4-8, del2-7, and del2-8. Each coding exon is colored, and noncoding exons are gray. The exons encoding the N-terminal DNA-binding, and C-terminal dimerization domains are indicated by arrows. (B) Expression levels of IK1 (ENST00000331340) in all B-ALL samples (including 99 diagnosis samples and matched relapse for 2 samples). Plot shows transcripts per million (TPM) on the y-axis. (C-D) Relationship between expression of the IKZF1 del 4-7 transcript and IKZF1 del 4-8 and total IKZF1 expression (the sum of all IKZF1 transcripts). Samples that have the respective IKZF1 deletion transcript are colored and all other samples are shown in gray. The legend for panels B-D is shown at top right. Samples are colored by IKZF1 deletion status based on primary and secondary RQ-PCR results, according to the key. Samples that have not been tested for IKZF1 deletion (unknown) or are confirmed nondeletions (undetected) are gray. Samples that were identified in primary RQ-PCR analysis, as part of clinical diagnostics (clinical RQ-PCR result) are depicted by circles, and samples that were predicted by RNA-seq and validated by secondary RQ-PCR (RNA-seq predicted and validated) are depicted by triangles. Samples that were predicted by RNA-seq to have an IKZF1 deletion but were not validated by RQ-PCR (unvalidated) are depicted by unfilled triangles.
Identifying IKZF1 deletions using RNA-seq data. (A) Schematic of the IKZF1 canonical transcript (IK1) and augmented deletion transcripts, del4-7, del4-8, del2-7, and del2-8. Each coding exon is colored, and noncoding exons are gray. The exons encoding the N-terminal DNA-binding, and C-terminal dimerization domains are indicated by arrows. (B) Expression levels of IK1 (ENST00000331340) in all B-ALL samples (including 99 diagnosis samples and matched relapse for 2 samples). Plot shows transcripts per million (TPM) on the y-axis. (C-D) Relationship between expression of the IKZF1 del 4-7 transcript and IKZF1 del 4-8 and total IKZF1 expression (the sum of all IKZF1 transcripts). Samples that have the respective IKZF1 deletion transcript are colored and all other samples are shown in gray. The legend for panels B-D is shown at top right. Samples are colored by IKZF1 deletion status based on primary and secondary RQ-PCR results, according to the key. Samples that have not been tested for IKZF1 deletion (unknown) or are confirmed nondeletions (undetected) are gray. Samples that were identified in primary RQ-PCR analysis, as part of clinical diagnostics (clinical RQ-PCR result) are depicted by circles, and samples that were predicted by RNA-seq and validated by secondary RQ-PCR (RNA-seq predicted and validated) are depicted by triangles. Samples that were predicted by RNA-seq to have an IKZF1 deletion but were not validated by RQ-PCR (unvalidated) are depicted by unfilled triangles.
First, we showed that lower expression levels of IKZF1 were not indicative of IKZF1 deletion (Figure 4B). We next augmented our reference transcriptome to include IKZF1-deleted sequences (detailed in “Materials and methods”). We included additional transcripts describing IKZF1 del 4-7 (IKZF1 deletion of exons 4-7), IKZF1 del2-7, IKZF1 del2-8, and IKZF1 del 4-8 (Figure 4A). We estimated the abundance of each deletion transcript using Salmon28 and plotted the result against the total number of IKZF1 transcripts (Figure 4C-D; supplemental Figure 5). We observed elevated expression of the IKZF1 del4-7 transcript in the 3 samples with known IKZF1 del4-7. In addition, we detected IKZF1 del 4-7 in 5 untested cases and confirmed the deletion in all of them by RQ-PCR (Figure 4C). One sample (B-ALL_10) was known to have an IKZF1 del4-7 at relapse, and RNA-seq analysis detected the presence of a deletion in the diagnostic sample. B-ALL8_6 and B-ALL7_5 were known to harbor IKZF1 del4-7 deletions at relapse. RQ-PCR, but not RNA-seq, confirmed the presence of subclonal deletions at diagnosis (supplemental Figure 6).
The relationships between the estimated abundance of the other deletion transcripts and the total abundance of the IKZF1 transcript was less clear, probably because of the greater uncertainty in the estimations of transcript abundance (Figure 4D; supplemental Figure 5). RNA-seq predicted the presence of an IKZF1 del4-8 in a single sample (B-ALL7_8; Figure 4D). However, the RNA-seq prediction of IKZF1 del4-8 is not reliable using this method. Although IKZF1 deleted samples tended to have a lower proportion of the canonical IK1 transcript, compared with the total number of estimated transcripts, this alone did not reliably predict IKZF1 intragenic deletion (supplemental Figure 7). Interestingly, 1 sample expressed 2 out-of-frame IKZF1 fusion transcripts that disrupt IKZF1 expression (supplemental Figure 8), identified only by RNA-seq (B-ALL11_12). Supplemental Table 6 summarizes RQ-PCR results from primary (clinical) and secondary analyses. Together these data show that RNA-seq can be used to detect the most common IKZF1 intragenic deletion.
AllSorts, a gene expression classifier to identify Ph-like, ETV6-RUNX1+, and ERG-deleted/DUX4-rearranged subtypes
We built a gene expression classifier to identify ETV6-RUNX1+ (ETV), Ph-like, and ERG-deleted/DUX4-rearranged (ERG) B-ALL, using a random forest classifier of RNA-Seq expression data (https://github.com/Oshlack/AllSorts). We applied the classifier to all 99 B-ALL samples (supplemental Figure 9). One hyperdiploid sample, with an IKZF1 deletion, was classified as ERG (B-ALL6_2); however, the presence of a DUX4 rearrangement could not be confirmed. We assessed how accurately the classifier predicted the class of samples known to harbor Ph-like and ETV6-RUNX1 rearrangements (Figure 5). Eighteen of 20 ETV6-RUNX1+ B-ALL cases were correctly predicted. One of 4 nonstandard ETV6 rearrangement cases (ETV6 other) classified as ETV6-RUNX1+, expressing an out-of-frame ETV6-NR3C1 fusion and no IKZF1 deletion (B-ALL18_1). ETV6-RUNX1-like is a more recently classified subtype of B-ALL, defined by an ETV6-RUNX1-like gene expression profile and coexisting ETV6 and IKZF1 alterations.20
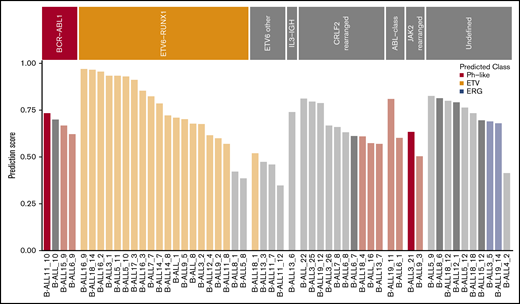
Using a B-ALL gene expression classifier to identify Ph-like, ETV6-RUNX1+, and ERG-deleted/DUX4-rearranged cases. Bar chart of prediction probabilities for B-ALL samples grouped by molecular subtype BCR-ABL1, ETV6-RUNX1, ETV6 other, IL3-IGH, CRLF2-rearranged, ABL-class, JAK2-rearranged, or undefined. Samples were classified into Ph-like (red), ETV6-RUNX1+(orange), or ERG-deleted (blue) if they received a prediction score over 0.5 for any of these classes. Samples that were classified as other with a prediction score >0.75 or were not classified are gray. Bars are dark colored for samples where the patient either relapsed or died and light colored for all other cases.
Using a B-ALL gene expression classifier to identify Ph-like, ETV6-RUNX1+, and ERG-deleted/DUX4-rearranged cases. Bar chart of prediction probabilities for B-ALL samples grouped by molecular subtype BCR-ABL1, ETV6-RUNX1, ETV6 other, IL3-IGH, CRLF2-rearranged, ABL-class, JAK2-rearranged, or undefined. Samples were classified into Ph-like (red), ETV6-RUNX1+(orange), or ERG-deleted (blue) if they received a prediction score over 0.5 for any of these classes. Samples that were classified as other with a prediction score >0.75 or were not classified are gray. Bars are dark colored for samples where the patient either relapsed or died and light colored for all other cases.
We identified a Ph-like gene expression signature in 3 of 10 (30%) CRLF2-rearranged B-ALL cases, 2 of which had coexisting IKZF1 deletions and 1 of which had a PAX5 rearrangement. The remaining CRLF2 fusions were classified as “other,” even if a coexisting IKZF1 deletion was present (supplemental Table 7). The classifier also identified a Ph-like gene expression profile in all ABL-class and JAK2-rearranged cases, as expected.7 One sample, known to have a BCR-ABL1 fusion, but a very low tumor burden (B-ALL_10) fell below the probability cutoff (0.5) for Ph+ ALL.
The classifier was applied to the 10 B-ALL samples, with no clinically relevant fusions detected by RNA-seq and no positive standard molecular test (supplemental Table 8). Two samples were predicted to be ERG (B-ALL3_5 and B-ALL19_14) and expressed the highest DUX4 (supplemental Figure 10). Manual inspection of aligned reads in the integrative Genomics Viewer identified an IGH-DUX4 rearrangement in 1 of these samples (supplemental Figure 11). This sample also had high PDGFRA expression, a recurring feature of ERG-deleted/DUX4-rearranged ALL (supplemental Figure 10).37 In all, the random forest classifier was useful in further refining the subtype classification of this cohort of ALL; however, a tool trained on a larger and more diverse set of samples will have greater clinical utility.
Discussion
RNA-seq is a well-established technology for identification of fusion transcripts and gene expression profiling in a research setting, but is currently not used in clinical diagnostics for ALL in Australia.7,20,24 In this study, we have assessed the utility of RNA-seq to identify genomic features of ALL in a cohort of 126 patients. In addition, we have used RNA-seq data to identify IKZF1 deletions and have developed a preliminary gene expression classifier to identify ETV6-RUNX1+, ERG-deleted/DUX4-rearranged, and Ph-like B-ALL.
In our cohort, RNA-seq detected 86% of fusion transcripts that were captured by standard-of-care diagnostics. Although RNA-seq and fusion finding algorithms are widely used for fusion detection, there are limitations to this approach. Our data, together with the work of others, show that RNA-seq may not detect fusions in samples with low tumor burdens or that are expressed at low levels (such as KMT2A rearrangements). Increasing the depth of sequencing, albeit at greater cost per sample, could diminish this problem. However, a more inherent limitation of RNA-seq is the detection of rearrangements involving promotor and enhancer regions (such as IGH rearrangements).20,38 Although RNA-seq does not directly identify such fusions, overexpression of a target gene could be used as a surrogate measure to identify these rearrangements, as seen in 2 of our patients with IL3-IGH and IGH-CRLF2 fusions.39,40
A significant advantage of an unbiased RNA-seq approach, as distinct from targeted sequencing, is the capacity to identify unexpected or previously unknown fusions that are potentially clinically actionable. We identified 2 PAX5-JAK2 rearrangements and a TCF7-CSF1R rearrangement in a T-ALL patient, which would be predicted to be sensitive to ruxolitinib and dasatinib, respectively.33 Both fusions are the result of intrachromosomal deletions and were not detected clinically by conventional karyotype analysis. In addition, FISH is not routinely used to test for rearrangements involving JAK2 or CSF1R. Further, a fusion such as ETV6-CRX suggests some intriguing biology and prompts further investigation to determine the mechanisms by which the enforced overexpression of the CRX homeodomain may block T-cell differentiation. However, in cases where standard-of-care testing definitively identifies a known ALL subtype, RNA-seq, at least in our hands, offered little further information. Therefore, we posit that RNA-seq will have the most significant clinical impact in patients who are not classified into an established subtype, specifically in B-other or T-ALL.
We have shown that RNA-seq data can be used to detect the most common intragenic IKZF1 deletion. This methodology relied on measuring the abundance of the specific transcript associated with IKZF1 del4-7 deletions. The detection of less common IKZF1 deletions was not reliable; however, this is not likely to be an inherent limitation of the approach, but more a lack of certainty in the counts of other IKZF1 deletion-specific transcripts. Larger cohorts and a clearer picture of the relationship between other IKZF1 deletions and the transcripts they generate would allow the detection of the full range of IKZF1 deletions.
The B-ALL gene expression classifier, although not dissimilar in principal to other published techniques that detect Ph+ or Ph-like ALL,41 uses RNA-seq to measure gene abundance and can also call the probability of other B-ALL subtypes. With this tool, 2 ERG-deleted/DUX4-rearranged cases were identified that had not been picked up by standard-of-care testing or fusion calling.24,42 It is evident from the expression analysis of large ALL cohorts, that an ALL subtype classifier based on gene expression will have significant power to detect a much larger range of ALL entities.20
Comprehensive diagnostic pipelines integrating next-generation sequencing techniques, including transcriptome, whole-genome, and whole-exome sequencing, have been implemented in larger research hospitals, but the utility of this approach in a smaller context has not been established.43 We have shown that even a single approach like RNA-seq can identify many additional molecular features in ALL that have potential to be clinically actionable. Even when the limitations of RNA-seq are taken into consideration, we conclude that additional clinical benefit can be gained from implementing RNA-seq pipelines, specifically for patients who do not have a standard lesion identified by standard-of-care diagnostics.
The data reported in this article have been submitted to the European Genome-Phenome Archive (accession number EGAS00001004212).
Acknowledgments
The authors thank the Research Genomics Facility of the Victorian Clinical Genetics Service.
The RQ-PCR testing was supported by Cancer Australia PdCCRs APP1128727 Sutton. The establishment and running of the Children’s Cancer Centre Tissue Bank are made possible through the generous support of Cancer In Kids@RCH (www.cika.org.au), Leukaemia Auxiliary at RCH (LARCH), the Murdoch Children’s Research Institute, and The Royal Children’s Hospital Foundation. This work was supported by the Victorian Government’s Operational Infrastructure Support Program; the Children’s Cancer Foundation (Project 131 and 132); National Health and Medical Research Council grant 1140626; SCOR grant 7015-18 from the Lymphoma and Leukemia Society; and an Australian Government Research Training Program Scholarship (L.M.B.); and the Victorian Government’s Operational Infrastructure Support Program. S.L.K. is supported by a fellowship from the Victorian Cancer Agency and I.J.M. is supported by a fellowship from the Victorian Cancer Agency and by the Felton Bequest.
Authorship
Contribution: L.M.B. contributed to the conceptual framework of the manuscript, RNA sequencing, data analysis, figure preparation, and wrote the first draft of the manuscript; A.L. performed bioinformatics analysis, including the IKZF1 analysis, and prepared the figures; A.Z. and E.W collected and collated patient clinical information and assisted in interpretation of clinical data; N.M.D. and B.S. assisted in bioinformatics analysis; A.H. developed AllSorts gene expression classifier; M.M., F.M.M., and S.L.K. provided clinical information and clinical perspectives of data interpretation; R.C.B. contributed to RNA sequencing; L.E.A.L. managed the identification, access, and provision of samples for sequencing; J.C., I.B., and V.P. performed karyotyping, FISH, and SNP microarray analysis; N.C.V. and R.S. generated the IKZF1 RQ-PCR data; I.J.M. provided intellectual input to the study and manuscript; A.O. and P.G.E. conceived and supervised the study and wrote the final version of the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paul G. Ekert, Murdoch Children’s Research Institute, Flemington Rd, Parkville, VIC 3052, Australia; e-mail: paul.ekert@mcri.edu.au; or Alicia Oshlack, Murdoch Children’s Research Institute, Flemington Rd, Parkville, VIC 3052, Australia; e-mail: alicia.oshlack@petermac.org.

