

Key Points
CCT241736 is a dual inhibitor of FLT3 and Aurora kinases with significant activity in vivo and in quizartinib-resistant patient samples.
CCT241736, with its unique kinase inhibitory profile, is a candidate for FLT3-ITD and FLT3-TKD AML patients with resistance to current drugs.

Abstract
Internal tandem duplication of FLT3 (FLT3-ITD) is one of the most common somatic mutations in acute myeloid leukemia (AML); it causes constitutive activation of FLT3 kinase and is associated with high relapse rates and poor survival. Small-molecule inhibition of FLT3 represents an attractive therapeutic strategy for this subtype of AML, although resistance from secondary FLT3 tyrosine kinase domain (FLT3-TKD) mutations is an emerging clinical problem. CCT241736 is an orally bioavailable, selective, and potent dual inhibitor of FLT3 and Aurora kinases. FLT3-ITD+ cells with secondary FLT3-TKD mutations have high in vitro relative resistance to the FLT3 inhibitors quizartinib and sorafenib, but not to CCT241736. The mechanism of action of CCT241736 results in significant in vivo efficacy, with inhibition of tumor growth observed in efficacy studies in FLT3-ITD and FLT3-ITD-TKD human tumor xenograft models. The efficacy of CCT241736 was also confirmed in primary samples from AML patients, including those with quizartinib-resistant disease, which induces apoptosis through inhibition of both FLT3 and Aurora kinases. The unique combination of CCT241736 properties based on robust potency, dual selectivity, and significant in vivo activity indicate that CCT241736 is a bona fide clinical drug candidate for FLT3-ITD and TKD AML patients with resistance to current drugs.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematopoietic malignancy that is associated with poor outcomes for many patients.1 FLT3 is a class 3 receptor tyrosine kinase, a class that includes platelet-derived growth factor receptor α and β, c-KIT, and c-fms.2 It is expressed in a variety of human and murine cell lines of both myeloid and B-lymphoid lineage. When activated by its cognate FLT3 ligand (FL), it initiates downstream signaling via the phosphatidylinositol 3-kinase/AKT/mTOR pathway. FLT3 also activates the Ras/Raf/MEK/ERK pathway via Src homology and collagen (SHC) proteins and GRB2-binding protein (GAB2), which leads to downstream activation of cyclic adenosine monophosphate response element-binding protein (CREB) and signal transducer and activator of transcription 5 (STAT5).3,4 Mutation of the FLT3 gene is a frequent event in AML and usually involves internal tandem duplication (ITD) of the juxtamembrane domain coding region or point mutations of the tyrosine kinase domain (TKD).5 Both FLT3-ITD and FLT3-TKD mutations result in ligand-independent proliferation because of the constitutive dimerization and activation of the FLT3 receptor.5
Several small molecule kinase inhibitors targeting FLT3 have been evaluated in AML patients, but the clinical impact has thus far been limited because of the transient responses when they are used as single agents and the emergence of acquired resistance after treatment.6 One particular mechanism of resistance is acquired secondary mutations in FLT3-TKD. All 9 patients analyzed from the phase 2 study of AC220 (quizartinib) who relapsed after achieving complete bone marrow responses had secondary FLT3-TKD mutations (at residues F691 or D835) on the FLT3-ITD+ allele.7 Similarly, secondary FLT3-TKD mutations at D835 have been identified in FLT3-ITD patients relapsing after initially successful therapy with sorafenib. Importantly, evidence suggests that double FLT3-ITD-TKD mutations are present in treatment-naïve, leukemia-initiating subclones and are selected out by therapy with sorafenib.8 In addition to secondary FLT3-TKD mutations, increased levels of FL have also been demonstrated to cause relative resistance to FLT3 inhibitors.9
Aurora kinases are a family of highly conserved serine-threonine protein kinases that play a key role in several stages of mitosis.10 An increase in the expression of Aurora A is associated with poor prognosis in a range of cancers, including leukemia in which overexpression of Aurora A has been consistently demonstrated.11 Aurora A inhibition by alisertib has been shown to arrest cells in the G2-M phase in a dose- and time-dependent manner.12 AML patients express a disproportionate number of undifferentiated cells (CD34+/CD38–), which aberrantly express greater levels of Aurora A than their CD34+/CD38+ counterparts. This undifferentiated population shows greater sensitivity to alisertib.13 Alisertib has been demonstrated to have modest single-agent activity in a phase 2 clinical trial (NCT00830518) in AML patients. Although 17% of patients showed a complete or partial response and 49% showed stable disease, there was no causal link between disease subtype and progression and response rate.14
Here, we report the preclinical activity of CCT241736, a potent, orally bioavailable, dual FLT3-Aurora kinase inhibitor.15 In both in vitro and in vivo models, we demonstrated that the presence of secondary TKD mutations (D835Y and F691L) in a human FLT3-ITD+ AML cell line or exposure to increased levels of FL result in high relative resistance to the FLT3 inhibitor AC220 but not to CCT241736. In addition, CCT241736 induces apoptosis in primary samples from patients with AML and those with quizartinib-resistant AML independently of their FLT3 status because of its dual inhibitory activity.
Materials and methods
Cell culture
The human AML cell lines MOLM-13 and MV4-11 were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ). The DSMZ authenticates all human cell lines by DNA typing and confirms species of origin by polymerase chain reaction analysis. Working stocks for the experiments described in this study were prepared immediately upon receipt of cells from DSMZ. Both cell lines are FLT3-ITD+16 and were maintained in antibiotic-free RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; PAA Laboratories Ltd, Somerset, United Kingdom). MOLM-13 cells with resistance to the FLT3 inhibitor MLN518 (tandutinib; hereafter MOLM-13-RES) were developed by culturing MOLM-13 cells in the presence of increasing concentrations of MLN518 (with ≤0.1% dimethyl sulfoxide [DMSO]) until confluent growth was sustained in 5 μM MLN518 as described previously.17 Experiments using MOLM-13-RES cells were carried out after at least overnight incubation in MLN518-free RPMI 1640 medium with 10% FBS. The same method was used to generate the MOLM-13-RES-AC cell line by using increasing concentrations of AC220 up to approximately 1 μM.
Compounds
CCT241736 and CCT137690 were discovered and synthesized at our Institute,15,18 and MLN518 and sorafenib were purchased from LC Laboratories (Woburn, MA). AC220 was purchased from Activate Scientific GmbH (Prien, Germany). Nocodazole and cytarabine were purchased from Sigma-Aldrich (St. Louis, MO). All compounds were dissolved in DMSO and stored at −20°C.
In vitro kinase assays
The concentration of the compound that inhibited FLT3 and FLT3 (D835Y) kinase activity by 50% of normal (IC50) was determined by Z′-LYTE assay using Invitrogen’s SelectScreen Biochemical Kinase Profiling Service (Invitrogen, Paisley, United Kingdom). The adenosine triphosphate concentration used for these assays was equal to Km apparent.
Cellular assays
To assess cell viability in vitro, all cell lines were seeded into 96-well plates at a density of 2 × 105 cells per 100 μL and then treated with 0.2% DMSO or varying concentrations of CCT241736, CCT137690, or MLN518 for 72 hours. Cell viability was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium [MTS]; Promega, Madison, WI) with absorbance measured at 490 nm using a Wallac Victor2 1420 multilabel counter (PerkinElmer Life Sciences, Cambridgeshire, United Kingdom). For cellular assays to assess inhibition of kinase signaling pathways and apoptosis, cell lines were seeded at varying concentrations in 12- or 24-well plates or 25 cm2 flasks, respectively.
Immunoblotting
Protein extraction was performed as previously described,17 and the extracted proteins were immunoblotted with the following antibodies: phospho-Aurora-A (Thr288)/Aurora-B (Thr232)/Aurora-C (Thr198), Aurora-A, phospho-FLT3 (Tyr 842), phospho-STAT5 (Tyr694), STAT5, phospho-p44/42 MAPK (Thr202/Tyr204), p44/42 MAPK, and poly (ADP-ribose) polymerase (PARP) (all from Cell Signaling Technology, Danvers, MA); FLT3 (C-20, Santa Cruz Biotechnology, Santa Cruz, CA); Aurora-B and histone H3 (HH3) (both from Abcam); and phospho-histone H3 (Ser10) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (both from Millipore, Billerica, MA). For detection of phosphorylated FLT3 in xenograft samples, cell lysates were immunoprecipitated with FLT3 antibody and protein A sepharose (GE Healthcare, Buckinghamshire, United Kingdom), resolved by electrophoresis, immunoblotted onto polyvinylidene fluoride membranes (Millipore, Billerica, MA), and probed with a phospho-tyrosine (4G10) antibody (Millipore). After exposure, membranes were stripped with 5% acetic acid and then re-probed with the total FLT3 antibody. All membranes were incubated with secondary anti-rabbit or anti-mouse antibodies conjugated to horseradish peroxidase (Dako, Glostrup, Denmark), developed by using enhanced chemiluminescence substrate (Thermo Fisher Scientific, Rockford, IL) and exposed on enhanced chemiluminescence Amersham Hyperfilm (GE Healthcare). Apoptosis was examined by detection of PARP cleavage using immunoblotting.
Cell cycle analysis
Cells were fixed overnight at 4°C in 70% ethanol, washed in phosphate-buffered saline (PBS) (with 1% FBS), and then incubated for 30 minutes in PBS with 1% FBS, 0.04% propidium iodide, and 0.25% RNase. Cells were analyzed using a BD LSRII flow cytometer (BD Biosciences).
Patient samples
Peripheral blood samples were obtained from newly diagnosed AML and quizartinib-resistant AML patients at Royal Marsden Hospital. After written informed consent was provided, the study was approved by the East of England-Cambridge South Research Ethics Committee. All studies complied with the rules of the Review Board and the revised Helsinki protocol. The samples were collected at untreated presentation or relapse, and mononuclear cells were obtained by density gradient centrifugation. Patients with available cytogenetic data were classified according to the Medical Research Council classification system.19
Primary cell assays
Mouse stromal cell line MS-5 obtained from DSMZ (Braunschweig, Germany) was used to support human AML primary cells. MS-5 cells were irradiated with 7.5 Gy using a gamma irradiator and cultured in Minimal Essential Medium alpha supplemented with 10% heat-inactivated FBS and penicillin-streptomycin in 12-well plates at a density of 5 × 104 cells per milliliter in a humidified incubator containing 5% CO2 at 37°C. After 24 hours, AML cells were cocultured in triplicate onto irradiated MS-5 cells at a density of 1.2 million cells per milliliter with 3 different doses of FLT3 inhibitor. Three days after treatment, cells were harvested and counted using a Luna-II Automated Cell Counter (Labtech). Annexin V and Vybrant FAM caspase-3 and caspase-7 (caspase-3/-7) assays were also used to assess apoptosis.
Apoptotic assays
For annexin V assays, cells were washed using annexin V binding buffer (BD Biosciences), suspended at a concentration of 1 × 106 cells per milliliter, and incubated with annexin V-fluorescein isothiocyanate and human CD45-allophycocyanin simultaneously for 15 minutes at room temperature in the dark. Cells were then washed and resuspended in PBS with 2% fetal calf serum and 4′,6-diamidino-2-phenylindole (DAPI) and analyzed on a BD FACSCanto II cytometer. Annexin V–positive cells (both DAPI negative and DAPI positive) were defined as apoptotic cells. Caspase-3/-7 activity was measured using the Vybrant FAM caspase-3/-7 assay according to the manufacturer’s instructions (Invitrogen). Cells were analyzed on a BD FACSCanto II cytometer and analyzed with FlowJo software.
Animal efficacy studies
All animal studies were approved by the local research ethics committee and were carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and national guidelines.20 Athymic CrTac:NCr-Fox1 (nu) mice were bred in-house. Female mice 6 to 8 weeks old were injected subcutaneously in the right flank with 2 × 106 MOLM-13 or MOLM-13-RES cells. When mean tumor diameter was 6 mm (at ∼day 5), mice were assigned to treatment or control cohorts (8 mice each), and dosing began with the vehicle, CCT241736, MLN518, or AC220 at the indicated doses. Tumors were routinely measured across 2 perpendicular diameters, and volumes were calculated by using the formula V = 4/3π [(d1 + d2)/4].3 Cohorts of mice were culled at specified times after the final dose, and tumors were excised, weighed, measured, and processed for pharmacokinetic (PK) and pharmacodynamic (PD) analyses. For the systemic model, 2 × 105 BaF3FLT3-ITD F691L luciferase-expressing cells were injected into NOD SCID mice. Mice were dosed with vehicle control 4 days after tumor cell implantation and randomization. Half the mice received a combination of DMSO (10%), Tween 80 (5%), PEG 400 (20%), and water (65%) and half received hydroxypropyl β cyclodextrin (22%), AC220 5 mg/kg orally once per day, and CCT241736 100 mg/kg orally twice per day; tumor burden was then assessed by whole-body bioluminescent imaging. Animals were culled when they showed signs of deterioration due to tumor burden (body weight loss, rapid breathing).
Statistics
All statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software Inc, La Jolla, CA). In vitro log dose-response curves were calculated using nonlinear regression with variable slope after normalizing absorbance to untreated and cellular controls with the concentration required to inhibit the MTS response by 50% reported as the viability IC50. For in vivo studies, survival was calculated using the Kaplan-Meier method.
Results
In vitro activity, efficacy, and mechanism of action of CCT241736 in AML cell lines
We have shown that CCT241736 is a potent dual inhibitor of FLT3 and Aurora kinases with few off-target kinase activities across the human kinome.15 In addition, CCT241736 did not inhibit the major cytochrome P450 isoforms and hERG, with IC50 values greater than 10 μM. The PK profile of CCT241736 in mice and rats revealed a compound with high oral bioavailability, low clearance, and a moderate volume of distribution.15
To assess the cell-based activity of CCT241736 alongside the selective FLT3 inhibitor MLN518, we used FLT3-ITD+ MOLM-13 and MV4-11 AML cell lines together with KG-1a AML cell lines that were FLT3 wild-type (WT) (representative graphs in supplemental Figure 1A-C). In a 72-hour MTS proliferation assay, both CCT241736 and MLN518 potently inhibited the viability of both MOLM-13 (growth inhibition [GI50], 0.1 μM and 0.034 μM, respectively) and MV4-11 (GI50, 0.29 μM and 0.11 μM, respectively). CCT241736 but not MLN518 inhibited the viability of the KG-1a FLT3 WT cell line (GI50, 1 μM and >20 μM, respectively).
To confirm that the loss of cell viability in FLT3-ITD+ cells treated with dual FLT3-Aurora inhibitor was associated with apoptosis, MOLM-13 cells were treated with CCT241736 and analyzed for 2 different markers of apoptosis. Immunoblotting of cell lysates confirmed PARP cleavage as well as downregulation of survivin in a concentration-dependent manner at both 24 and 48 hours after treatment (Figure 1A). At a 0.5 μM concentration of CCT241736, there was robust cleavage of PARP at both time points and a complete loss of survivin expression, thus confirming that the loss of cell viability after treatment with CCT241736 was the result of induction of apoptosis.
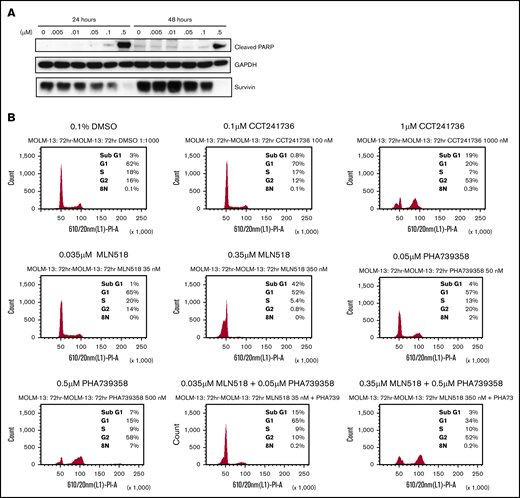
Induction of apoptosis, cell cycle regulation, and in vitro inhibition of FLT3 and Aurora signaling by CCT241736 in MOLM-13 cells. (A) Immunoblotting analysis of cells treated with CCT241736 at the indicated concentrations for 24 and 48 hours using antibodies specific for cleaved PARP and survivin. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Cell cycle profile of MOLM-13 cells treated with FLT3 and Aurora kinase inhibitors or their combinations: CCT241736, MLN518, PHA-739358, or MLN518 + PHA-739358. MOLM-13 cells were treated for 72 hours with the compounds at the indicated concentrations approximating their viability IC50 and 10 × IC50, and they were fixed, stained, and analyzed by fluorescence-activated cell sorting (FACS). Y-axes represent FACS event counts (same scale for all histograms). The percentage of cells in sub-G1, G1, S, G2, and 8N phases of the cell cycle are included. (C) After overnight incubation with 50 ng/mL nocodazole, MOLM-13 cells were treated with CCT241736, CCT137690, or MLN518 for 2.5 hours at the indicated concentrations. Cell lysates were prepared and analyzed for the expression of the indicated proteins by immunoblotting using specific antibodies. GAPDH was used as loading control.
Induction of apoptosis, cell cycle regulation, and in vitro inhibition of FLT3 and Aurora signaling by CCT241736 in MOLM-13 cells. (A) Immunoblotting analysis of cells treated with CCT241736 at the indicated concentrations for 24 and 48 hours using antibodies specific for cleaved PARP and survivin. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Cell cycle profile of MOLM-13 cells treated with FLT3 and Aurora kinase inhibitors or their combinations: CCT241736, MLN518, PHA-739358, or MLN518 + PHA-739358. MOLM-13 cells were treated for 72 hours with the compounds at the indicated concentrations approximating their viability IC50 and 10 × IC50, and they were fixed, stained, and analyzed by fluorescence-activated cell sorting (FACS). Y-axes represent FACS event counts (same scale for all histograms). The percentage of cells in sub-G1, G1, S, G2, and 8N phases of the cell cycle are included. (C) After overnight incubation with 50 ng/mL nocodazole, MOLM-13 cells were treated with CCT241736, CCT137690, or MLN518 for 2.5 hours at the indicated concentrations. Cell lysates were prepared and analyzed for the expression of the indicated proteins by immunoblotting using specific antibodies. GAPDH was used as loading control.
Inhibition of Aurora A kinase classically results in temporary G2-M arrest and cytokinesis failure followed by cell death, whereas Aurora B inhibition causes polyploidy due to abrogation of the mitotic checkpoint, failure of cytokinesis, and endoreduplication followed by cell death at longer treatments (>48 hours).21 FLT3 inhibition with compounds such as CEP-701 and FI-700 has been shown to cause G1 arrest, with a decrease in the S and G2-M populations. To determine whether the cell cycle effects of dual FLT3-Aurora inhibition were different from selective inhibition of either kinase alone, MOLM-13 cells were treated for 72 hours with CCT241736 (dual FLT3-Aurora), MLN518 (FLT3 selective), PHA-739358 (a pan-Aurora kinase inhibitor), and a combination of MLN518 and PHA-739358 (Figure 1B). Upon treatment with IC50 concentrations, there were no striking effects on the cell cycle profile for CCT241736 (0.1 μM) or MLN518 (0.035 μM). PHA-739358 caused polyploidy, with a small population of cells having 8N DNA content (0.05 μM). At concentrations 10-fold higher, MLN518 produced G1-S arrest, and CCT241736 caused both G1-S and G2-M arrest, whereas PHA-739358 caused increased polyploidy with a larger population of cells having 8N DNA content. It was notable that CCT241736 did not cause polyploidy. The phenotype of G1-S– and G2-M–arrested cells seen with CCT241736, distinct from the profile of the selective FLT3 inhibitor MLN518, was also seen in cells treated with a combination of MLN518 and PHA-739358, suggesting that in the setting of combined FLT3 and Aurora inhibition, polyploidy is abrogated. This particular profile mimics inhibition of both Aurora A and FLT3, consistent with the kinase profile of CCT241736 in biochemical assays. These data show the differences in mechanism of action between CCT241736 and the FLT3 selective (MLN518) or pan-Aurora (PHA-739358) inhibitors.
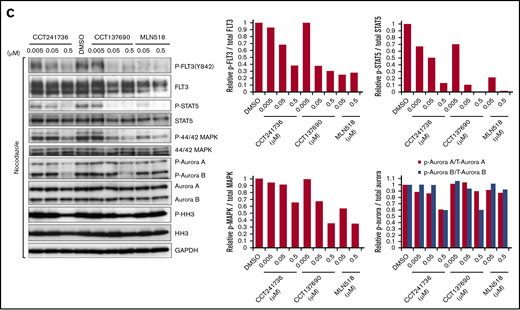
To assess biomarker modulation upon treatment with CCT241736, MOLM-13 cells were exposed to increasing concentrations of CCT241736, CCT137690 (an alternative FLT3-Aurora tool inhibitor compound with a slightly different selectivity profile of Aurora kinases compared with CCT241736),17,18 or MLN518. To more clearly assess the inhibition of endogenous Aurora A and B kinases, cells were incubated with nocodazole 50 ng/mL overnight to induce mitotic arrest, thus increasing the percentage of cells with high Aurora kinase expression. Immunoblotting was then performed on cell lysates to examine the inhibition of FLT3 (P-FLT3, P-STAT5, P-44/42-MAPK), and Aurora kinase signaling (P-Aurora A, P-Aurora B, P-HH3). In some cell lines, CCT137690 showed higher potency compared with CCT241736; however, the narrow safety margin against hERG of CCT137690 limited its further progression.18 Inhibition of FLT3 and downstream signaling was observed with all 3 compounds, whereas inhibition of Aurora kinase and downstream signaling was observed only with the 2 FLT3-Aurora inhibitors (Figure 1C; supplemental Figure 2).
Resistance to selective FLT3 inhibition
In view of the potency seen in vitro in MOLM-13 cells and the ability of CCT241736 to inhibit various FLT3-TKD mutations, it was hypothesized that dual FLT3-Aurora inhibitor CCT241736 may overcome resistance to selective FLT3 inhibitors such as MLN518 and AC220. Two important mechanisms of FLT3 inhibitor resistance were assessed: secondary FLT3-TKD mutations and increased FL levels. We have previously reported the generation of an AML cell line resistant to selective FLT3 inhibition through continuous culture of MOLM-13 cells in the presence of progressively increasing concentrations of MLN518.17 The resultant MOLM-13-RES cells had a new point mutation at D835, in addition to the original FLT3-ITD. As with other cell lines, the in vitro sensitivity of MOLM-13-RES cells to CCT241736 and MLN518 was assessed by MTS assay. CCT241736 potently inhibits WT and mutated FLT3 as well as the growth of MOLM-13 and MOLM-13-RES cells compared with MLN518: GI50 of CCT241736, 0.1 μM in MOLM-13 and 0.18 μM in MOLM-13-RES vs GI50 of MLN518, 0.03 μM in MOLM-13 and 3.57 μM in MOLM-13-RES. The biochemical activities of CCT241736 (Kd) were FLT3-WT, 0.006 μM; FLT3-ITD, 0.038 μM; and FLT3-D835Y, 0.014 μM. The biochemical activities for MLN518 were FLT3-WT, 0.003 μM; FLT3-ITD, 0.009 μM; and FLT3-D835Y, 1.8 μM. Although MLN518 potently inhibits wild-type and FLT3-ITD, it displays reduced activity in FLT3-D835Y and MOLM-13-RES cells (representative graphs in supplemental Figure 1D-E).
The inhibition of biomarkers downstream of FLT3 and Aurora kinase was examined in the FLT3 inhibitor–resistant cell line MOLM-13-RES in vitro. MOLM-13-RES cells were exposed to CCT241736 or CCT137690 or AMG-900 (a pan-Aurora inhibitor) for 4 hours at the indicated concentrations. Cells were then harvested and the levels of the downstream markers of FLT3 (P-STAT5) and Aurora kinase (phospho-histone H3 [P-HH3]) were evaluated. Levels of total STAT5 and total HH3 were used as controls. As expected, CCT241736 showed both FLT3 and Aurora kinase inhibition, MLN518 showed complete FLT3 inhibition only at high concentrations (5 μM) compared with CCT241736 and CCT137690 (0.5 μM), and AMG-900 showed Aurora inhibition with low FLT3 inhibition at high concentrations (Figure 2A). Although CCT137690 showed robust FLT3 pathway inhibition, it showed a weak P-HH3 inhibition. To more clearly assess the inhibition of endogenous Aurora A and B kinases, MOLM-13-RES cells were incubated with nocodazole to induce mitotic arrest. Immunoblotting was then performed on cell lysates to examine the inhibition of FLT3 pathway (P-STAT5) and Aurora kinases (P-Aurora A, P-Aurora B, P-HH3) by CCT137690 compared with MLN518. CCT137690 clearly inhibits both FLT3 pathway (P-STAT5) and Aurora kinases (P-Aurora A, P-Aurora B, and P-HH3), whereas MLN518 inhibits only the FLT3 pathway (supplemental Figure 2).
![Biomarker modulation by Aurora and FLT3 inhibitors in MOLM-13-RES cells. (A) MOLM-13-RES cells were arrested in mitosis by overnight incubation with 50 ng/mL nocodazole and treated with the indicated concentrations of CCT241736, CCT137690, MLN518, or AMG900 for 2.5 hours. Cell lysates were analyzed for STAT5 and HH3 phosphorylation by immunoblotting using specific antibodies. Total STAT5 and total HH3 were used as controls. The bar graphs (bottom panels) show compound cellular potency after normalization to the control total proteins. (B) BaF3FLT3-ITD and BaF3FLT3-ITD-F691L cells were treated for 2.5 hours with increasing concentrations of CCT241736 (20 nM to 10 μM) and with 50 nM AC220. Cell lysates were prepared, FLT3 was immunoprecipitated using a specific FLT3 antibody, and its phosphorylation was analyzed by immunoblotting with phospho-tyrosin–specific antibodies. The bar graph (bottom panel) shows CCT241736 and AC220 cellular potency after normalization to the control total proteins. (C) Fold change in viability IC50 with 10 ng/mL FL. MOLM-13 cells were treated with increasing concentrations of cytarabine, CCT241736, AC220, or MLN518 with or without 10 ng/mL FL for 72 hours. The IC50 values for viability were measured using MTS. Experiments were performed in parallel on 3 separate occasions. Error bars represent the mean change (± standard error of the mean [SEM]) in viability IC50 with 10 ng/mL FL compared with viability IC50 with no FL treatment. *P < .05 on paired Student t test analysis.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/7/10.1182_bloodadvances.2019000986/1/m_advancesadv2019000986f2.png?Expires=1765884406&Signature=Hi~PJVdqa~Nov2ET4cKwbmzcLgneCngh9C23MVPKxPW-by-kpAcl6Lbx75l5ObtE7wTJm7HwKCkDJy-GQzuupSIlWPhONyKuraUh5Fa68xFqMxkWaK-5ATPZjG7wNgToIQjwBzXW8pqwlr1WiDmd4LpxOBqKBf~a2Keiaf4EVT9kQioJXWURp4WtlZHnY5J1JqwXZmZUONV73J9c75C9JCaYle1rAyD4u~1tWOWVxJ3ttS7lLmLlpHoVv5ZxkMf1u807MzH~a-TsHdCmKxNzUj9nRuG4ez8y~m1ZWSaktgscVPKdiNp~U6OHru6O0qrvaXzA5scqsKBg1V52wSgVxA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Biomarker modulation by Aurora and FLT3 inhibitors in MOLM-13-RES cells. (A) MOLM-13-RES cells were arrested in mitosis by overnight incubation with 50 ng/mL nocodazole and treated with the indicated concentrations of CCT241736, CCT137690, MLN518, or AMG900 for 2.5 hours. Cell lysates were analyzed for STAT5 and HH3 phosphorylation by immunoblotting using specific antibodies. Total STAT5 and total HH3 were used as controls. The bar graphs (bottom panels) show compound cellular potency after normalization to the control total proteins. (B) BaF3FLT3-ITD and BaF3FLT3-ITD-F691L cells were treated for 2.5 hours with increasing concentrations of CCT241736 (20 nM to 10 μM) and with 50 nM AC220. Cell lysates were prepared, FLT3 was immunoprecipitated using a specific FLT3 antibody, and its phosphorylation was analyzed by immunoblotting with phospho-tyrosin–specific antibodies. The bar graph (bottom panel) shows CCT241736 and AC220 cellular potency after normalization to the control total proteins. (C) Fold change in viability IC50 with 10 ng/mL FL. MOLM-13 cells were treated with increasing concentrations of cytarabine, CCT241736, AC220, or MLN518 with or without 10 ng/mL FL for 72 hours. The IC50 values for viability were measured using MTS. Experiments were performed in parallel on 3 separate occasions. Error bars represent the mean change (± standard error of the mean [SEM]) in viability IC50 with 10 ng/mL FL compared with viability IC50 with no FL treatment. *P < .05 on paired Student t test analysis.
Biomarker modulation by Aurora and FLT3 inhibitors in MOLM-13-RES cells. (A) MOLM-13-RES cells were arrested in mitosis by overnight incubation with 50 ng/mL nocodazole and treated with the indicated concentrations of CCT241736, CCT137690, MLN518, or AMG900 for 2.5 hours. Cell lysates were analyzed for STAT5 and HH3 phosphorylation by immunoblotting using specific antibodies. Total STAT5 and total HH3 were used as controls. The bar graphs (bottom panels) show compound cellular potency after normalization to the control total proteins. (B) BaF3FLT3-ITD and BaF3FLT3-ITD-F691L cells were treated for 2.5 hours with increasing concentrations of CCT241736 (20 nM to 10 μM) and with 50 nM AC220. Cell lysates were prepared, FLT3 was immunoprecipitated using a specific FLT3 antibody, and its phosphorylation was analyzed by immunoblotting with phospho-tyrosin–specific antibodies. The bar graph (bottom panel) shows CCT241736 and AC220 cellular potency after normalization to the control total proteins. (C) Fold change in viability IC50 with 10 ng/mL FL. MOLM-13 cells were treated with increasing concentrations of cytarabine, CCT241736, AC220, or MLN518 with or without 10 ng/mL FL for 72 hours. The IC50 values for viability were measured using MTS. Experiments were performed in parallel on 3 separate occasions. Error bars represent the mean change (± standard error of the mean [SEM]) in viability IC50 with 10 ng/mL FL compared with viability IC50 with no FL treatment. *P < .05 on paired Student t test analysis.
To examine the potency of CCT241736 in other clinically relevant secondary FLT3-TKD mutations, we used a panel of BaF3 cells expressing FLT3-ITD or a range of clinically relevant FLT3-TKD mutants (Table 1; supplemental Figure 1F). Mutant FLT3-ITD or FLT3-TKD+ BaF3 cells have resistance to a wide range of FLT3 inhibitors (AC220, sorafenib, PLX3397, ponatinib, and DCC2036).7 Additional studies have further validated mutated FLT3 as an oncogenic driver and therapeutic target.7 Growth inhibition at CCT241736 concentrations <100 nM was observed against all FLT3 mutation–expressing cell lines studied (Table 1). Importantly, although the potent and selective FLT3 inhibitor AC220 displayed lower absolute GI50 values compared with CCT241736, the relative resistance of BaF3 cells with FLT3-TKD mutations to AC220 was 29- to 237-fold compared with cells with FLT3-ITD. In contrast, the relative resistance to CCT241736 was only 1.4- to 1.8-fold. The effect of CCT241736 on FLT3 phosphorylation was also examined in BaF3 cells engineered to express mutant FLT3-ITD+ or FLT3-ITD+-F691L. In both cell lines, CCT241736 produced a dose-dependent decrease in the phosphorylation of FLT3, which indicates robust potency in both mutants (Figure 2B).
Growth inhibition values of compounds in engineered BaF3 cell lines
| Compound | Parental cells | FLT3-ITD+ | F691L | Y842C | Y842H | D835V | D835Y | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | |||
| CCT241736 | 0.09 | 0.048 | 0.083 | 1.7 | 0.075 | 1.6 | 0.068 | 1.4 | 0.089 | 1.8 | 0.084 | 1.7 |
| AC220 | 1.4 | 0.0003 | 0.046 | 155 | 0.009 | 31 | 0.009 | 29 | 0.07 | 237 | 0.017 | 56 |
| Sorafenib | 4.3 | 0.0008 | 0.44 | 559 | 0.11 | 146 | 0.13 | 168 | 0.66 | 823 | 0.17 | 217 |
| Compound | Parental cells | FLT3-ITD+ | F691L | Y842C | Y842H | D835V | D835Y | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | GI50, μM | Fold of resistance | |||
| CCT241736 | 0.09 | 0.048 | 0.083 | 1.7 | 0.075 | 1.6 | 0.068 | 1.4 | 0.089 | 1.8 | 0.084 | 1.7 |
| AC220 | 1.4 | 0.0003 | 0.046 | 155 | 0.009 | 31 | 0.009 | 29 | 0.07 | 237 | 0.017 | 56 |
| Sorafenib | 4.3 | 0.0008 | 0.44 | 559 | 0.11 | 146 | 0.13 | 168 | 0.66 | 823 | 0.17 | 217 |
It has been shown that, despite constitutive FLT3 signaling, FLT3-ITD+ cells are further activated by FL. Endogenous FL expression promotes leukemic proliferation and reduces survival in FL knockout mouse models.22 The effect of exposure to 10 ng/mL FL on the in vitro efficacy of CCT241736 was therefore assessed in MOLM-13 cells and compared with MLN518 and AC220. Cytarabine was used as a nonspecific cytotoxic control. MLN518 and AC220 were most affected by high FL levels, whereas CCT241736 displayed no resistance to FL, in line with its relative selectivity and potency against both FLT3 and Aurora kinases (Figure 2C).
In vivo efficacy of CCT241736 in AML xenografts and in FLT3 inhibitor-resistant models
We tested whether the in vitro findings translated to the in vivo context. We carried out human tumor xenograft experiments (MOLM-13) in athymic mice to evaluate the efficacy of CCT241736 in vivo. CCT241736 reduced tumor growth compared with that in vehicle-treated mice in a dose-dependent manner with no observed toxicity on chronic dosing (Figure 3A).
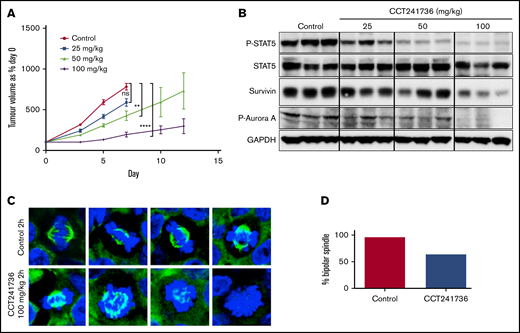
In vivo efficacy of CCT241736 in MOLM-13 human tumor xenografts. (A) Athymic mice (8 per cohort) were injected subcutaneously with 2 × 106 MOLM-13 cells. Five days after implantation, mean tumor diameter was 6 mm (day 0 on graph), and dosing began orally twice per day with vehicle, 25, 50, or 100 mg/kg. Tumor volumes were measured at days 0, 3, 5, 7, 10, and 12. Mean tumor volumes ± SEM are shown. Unpaired Student t test of 95% confidence intervals on day 7. ns, not significantly different (P = .14; control: CCT241736 at 25 mg/kg). **P = .08 (control: CCT241736 at 50 mg/kg); ****P < .0001 (control: CCT241736 at 100 mg/kg). (B) Biomarker modulation by CCT241736 in MOLM13 xenograft samples. Tumors were removed 2 hours after the final dose, and lysates were prepared. Equal amount of proteins from all the tumors were analyzed by immunoblotting using the indicated antibodies for P-STAT5 and Aurora signaling (P-Aurora A, survivin). GAPDH was used as loading control. (C) CCT241736 induces aberrant mitosis in MV4-11 xenograft tumors. Tumors from mice treated with 100 mg/kg CCT241736 for 18 days (orally twice per day) were removed 2 hours after the final dose and analyzed by immunofluorescence using α-tubulin (green) antibodies and DAPI (blue) (40× magnification). (D) Quantification of mitotic cells. At least 50 mitotic cells in control and treated tumors were counted for bipolar or aberrant mitosis.
In vivo efficacy of CCT241736 in MOLM-13 human tumor xenografts. (A) Athymic mice (8 per cohort) were injected subcutaneously with 2 × 106 MOLM-13 cells. Five days after implantation, mean tumor diameter was 6 mm (day 0 on graph), and dosing began orally twice per day with vehicle, 25, 50, or 100 mg/kg. Tumor volumes were measured at days 0, 3, 5, 7, 10, and 12. Mean tumor volumes ± SEM are shown. Unpaired Student t test of 95% confidence intervals on day 7. ns, not significantly different (P = .14; control: CCT241736 at 25 mg/kg). **P = .08 (control: CCT241736 at 50 mg/kg); ****P < .0001 (control: CCT241736 at 100 mg/kg). (B) Biomarker modulation by CCT241736 in MOLM13 xenograft samples. Tumors were removed 2 hours after the final dose, and lysates were prepared. Equal amount of proteins from all the tumors were analyzed by immunoblotting using the indicated antibodies for P-STAT5 and Aurora signaling (P-Aurora A, survivin). GAPDH was used as loading control. (C) CCT241736 induces aberrant mitosis in MV4-11 xenograft tumors. Tumors from mice treated with 100 mg/kg CCT241736 for 18 days (orally twice per day) were removed 2 hours after the final dose and analyzed by immunofluorescence using α-tubulin (green) antibodies and DAPI (blue) (40× magnification). (D) Quantification of mitotic cells. At least 50 mitotic cells in control and treated tumors were counted for bipolar or aberrant mitosis.
To analyze the PD activity of CCT241736 in vivo, tumors were collected at 2 hours after the final dose, and the lysates were analyzed by immunoblotting. Reduction of phospho-Aurora-A and phospho-STAT5 indicates inhibition of both Aurora A and FLT3 in a dose-dependent manner (Figure 3B). Similarly, survivin levels were reduced in a dose-dependent manner, which indicates that CCT241736 induced apoptosis in vivo. A confirmatory study in a second in vivo FLT3-ITD+ AML human tumor xenograft model (MV4-11) showed that CCT241736 significantly inhibits the growth of MV4-11 xenografts with clear inhibition of both HH3 and STAT5 phosphorylation at 2 hours after the final dose, indicating a dual inhibition of both Aurora and FLT3 downstream pathways in the tumor.15 To demonstrate that Aurora kinase inhibition by CCT241736 makes a significant contribution to efficacy in FLT3-mutated AML, samples of the MV4-11 tumors from the animals dosed with 100 mg/kg at 2 hours after administration were analyzed by immunofluorescence microscopy. More than one third (36%) of mitotic cells from tumors treated with CCT241736 formed monopolar or abnormal spindles, a typical phenotype for Aurora kinase inhibition.23 In comparison, this phenotype was observed in only 4% of mitotic cells in tumors from mice treated with vehicle control (Figure 3C-D). These data confirm the contribution of Aurora inhibition by CCT241736 to the in vivo efficacy and the already known induced phenotype from previous studies using Aurora inhibitors.23
The anti-tumor efficacy of CCT241736 and MLN518 were examined in an efficacy study using mouse xenografts of the MOLM-13-RES cell line. MOLM-13-RES cells remained sensitive to CCT241736, with an in vitro viability IC50 of 0.18 μM, which reflects a 1.8-fold difference in sensitivity compared with parental MOLM-13 cells. In contrast, the relative resistance of MOLM-13-RES cells to MLN518 was ∼100-fold (Table 1). In addition, cellular assays of MOLM-13-RES cells with CCT241736 showed a similar pattern of FLT3 and Aurora inhibition as in MOLM-13 cells (Figures 1C and 2A). To confirm the in vitro activity of CCT241736 against the MOLM-13-RES model of selective FLT3 inhibitor resistance, in vivo studies were performed using subcutaneous human tumor xenografts of MOLM-13-RES cells in athymic mice and the indicated compounds at their maximum tolerated doses (CCT241736 100 mg/kg twice per day; MLN518 160 mg/kg twice per day). As illustrated in Figure 4A, CCT241736 caused a significant in vivo growth inhibition in comparison with MLN518 (statistically significant difference was observed between MLN518 and CCT241736 but not between vehicle control and CCT241736 or MNL518, respectively). As in the parental MOLM-13 in vivo studies, biomarker modulation was consistent with inhibition of both FLT3 and Aurora kinases at well-tolerated doses (Figure 4B). In contrast, MLN518 caused no growth inhibition of MOLM-13-RES xenografts and no biomarker modulation or induction of apoptosis. The study had to be terminated when the control groups and the arm treated with MLN518 had reached the license volume limits.
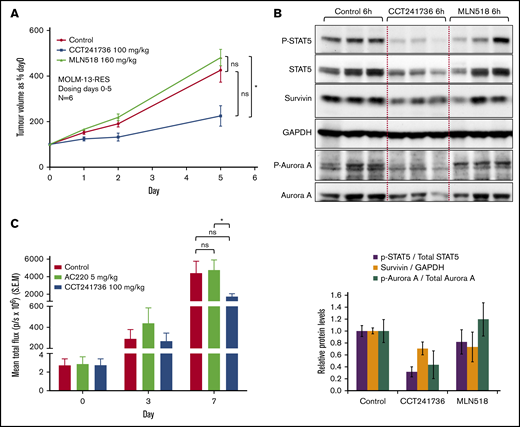
In vivo efficacy of CCT241736 in FLT3 inhibitor–resistant xenografts. (A) Athymic nude mice (6 per cohort) were injected subcutaneously with 2 × 106 MOLM-13-RES cells. Thirteen days after implantation, mean tumor diameter was 6 mm (day 0 on graphs), and dosing began orally twice per day for 5 days with solvent vehicle, CCT241736 at 100 mg/kg twice per day or MLN518 at 160 mg/kg twice per day. Tumor volumes were measured at days 0, 1, 2, and 5. Mean tumor volumes ± SEM are shown. Mice were culled at 1 and 6 hours after the final dose. Unpaired Student t test of 95% confidence intervals on day 5. ns, P = .13 (control: MLN518) or P = .4 (control: CCT241736). Significantly different, *P = .02 (MLN518:CCT241736). (B) Tumor lysates were analyzed by immunoblotting for expression of the indicated proteins. GAPDH, Aurora A, and STAT5 were used as controls. The bar graphs (bottom panel) show the in vivo potency of compounds after normalization to the control total proteins. (C) Systemic model of BaF3FLT3-ITD F691L luciferase–expressing tumors in NOD SCID mice. On day 7, the flux in the treated groups (mean total flux ± SEM) is indicated. Mice were dosed (starting 4 days after tumor cell implantation and randomization) with AC220 at 5 mg/kg orally once per day or CCT241736 at 100 mg/kg orally twice per day, and tumor burden was assessed by whole-body bioluminescent imaging. Animals were culled when they showed signs of deterioration as a result of tumor burden (body weight loss, rapid breathing). Unpaired Student t test of 95% confidence intervals on day 7. ns, P = .6 (control: AC220) or P = .1 (control: CCT241736). Significantly different, *P = .01 (AC220:CCT241736).
In vivo efficacy of CCT241736 in FLT3 inhibitor–resistant xenografts. (A) Athymic nude mice (6 per cohort) were injected subcutaneously with 2 × 106 MOLM-13-RES cells. Thirteen days after implantation, mean tumor diameter was 6 mm (day 0 on graphs), and dosing began orally twice per day for 5 days with solvent vehicle, CCT241736 at 100 mg/kg twice per day or MLN518 at 160 mg/kg twice per day. Tumor volumes were measured at days 0, 1, 2, and 5. Mean tumor volumes ± SEM are shown. Mice were culled at 1 and 6 hours after the final dose. Unpaired Student t test of 95% confidence intervals on day 5. ns, P = .13 (control: MLN518) or P = .4 (control: CCT241736). Significantly different, *P = .02 (MLN518:CCT241736). (B) Tumor lysates were analyzed by immunoblotting for expression of the indicated proteins. GAPDH, Aurora A, and STAT5 were used as controls. The bar graphs (bottom panel) show the in vivo potency of compounds after normalization to the control total proteins. (C) Systemic model of BaF3FLT3-ITD F691L luciferase–expressing tumors in NOD SCID mice. On day 7, the flux in the treated groups (mean total flux ± SEM) is indicated. Mice were dosed (starting 4 days after tumor cell implantation and randomization) with AC220 at 5 mg/kg orally once per day or CCT241736 at 100 mg/kg orally twice per day, and tumor burden was assessed by whole-body bioluminescent imaging. Animals were culled when they showed signs of deterioration as a result of tumor burden (body weight loss, rapid breathing). Unpaired Student t test of 95% confidence intervals on day 7. ns, P = .6 (control: AC220) or P = .1 (control: CCT241736). Significantly different, *P = .01 (AC220:CCT241736).
We also investigated the development of resistance to CCT241736 in vivo (dosing schedule and duration are presented in supplemental Figure 3A). MOLM-13 cells (1 × 107) were injected subcutaneously into female athymic mice. Once tumors were established (4 days), groups of 6 mice were administered vehicle control or CCT241736 100 mg/kg twice per day. On day 8, control mice were culled because of tumor size. By day 13, a tumor in 1 mouse had regressed, and the mouse was taken off the drug for 13 days to allow for regrowth; 2 other mice were taken off the drug for 2 days to allow for tumor regrowth. On day 24, the tumor of 1 of these latter mice was excised and transplanted as solid fragments into 8 mice.
Dosing of these mice commenced on day 35 at 100 mg/kg twice per day and continued to day 53 (17 days). At this point, tumors from 2 mice were excised and used as solid explants in 8 additional mice each. Other tumors were taken for in vitro sensitivity testing at this point (passage 2). Dosing of 4 mice from each group began on day 67 at 100 mg/kg twice per day and continued to day 73 (6 days). Tumors were then allowed to grow for 7 days off therapy (day 80) before 1 tumor was excised and used to provide solid explants in 8 additional mice. Animals were allowed to recover for 17 days (day 91), at which point dosing began at 150 mg/kg twice per day. After 5 days, the dose was reduced to 150 mg/kg once per day because of weight loss in the mice. Animals were finally culled after 13 days of dosing (day 107). The growth inhibition IC50’s were estimated for both CCT241736 and MLN518 in 2 lines established from the tumors taken at passage 2 and 2 lines taken at passage 4 and compared with the parental MOLM-13 cell line. Effectively, no change was observed in the IC50 of any cell line to either CCT241736 or the MLN518 (supplemental Figure 3B-C).
By using an in vitro saturation mutagenesis assay, AC220 resistance-conferring mutations were identified and reported at 4 residues in the kinase domain of FLT3-ITD.7 Mutation at 1 of these residues (the gatekeeper residue [F691]) has also been identified in AC220-relapsed patients. This substitution conferred a high degree of resistance to AC220 and was also cross-resistant to sorafenib in cell-based growth and biochemical assays.7 These data suggest that substitutions at F691 in FLT3-ITD will pose substantial barriers to disease control in AML patients treated with either AC220 or sorafenib; therefore, they represent high-value targets for efforts to develop novel FLT3 inhibitors. To examine the sensitivity of FLT3-ITD-F691L–resistant mutants to CCT241736, we performed in vivo studies using a model to simulate AML with the clinically relevant gatekeeper dual FLT3-ITD-F691L–resistant mutant. SCID mice injected intravenously with FLT3-ITD-F691L luciferase-expressing BaF3 cells were imaged to detect tumor establishment on day 4 and were randomly assigned to treatment groups: vehicle control, CCT241736 at the maximum tolerated dose of 100 mg/kg twice per day and AC220 at the maximum tolerated dose of 5 mg/kg once per day. Animals were imaged during treatment and culled when the systemic disease was evident in livers and spleens. On day 7 after the treatment, initiation of the total flux (protons/s) as a surrogate for tumor burden in bioluminescence imaging (expressed as percent of vehicle control) was as follows: AC220, 105%, and CCT241736, 42%, which confirmed that in this model there was a significant benefit of CCT241736 treatment in reducing tumor growth when compared with the FLT3 selective inhibitor AC220 (Figure 4C; supplemental Figure 4). Statistically significant difference was observed between AC220 and CCT241736.
CCT241736 potency in primary AML cells
To investigate whether CCT241736 has activity in primary AML cells, we obtained peripheral blood blasts from quizartinib-responded (13 samples: AML-1 to AML-13; supplemental Table 1) and quizartinib-relapsed (3 samples: AML-7, AML-14, and AML-15; Table 2 in bold) patients with diverse molecular characteristics, including those with FLT3 WT, FLT3-ITD, IDH1/2, and NPM1 mutations.24,25 AML cells from quizartinib-responded patients (AML-1 to AML-13) were cocultured in triplicate onto irradiated MS-5 fibroblasts and treated with 3 different concentrations of CCT241736 (0.3, 1, and 3 μM). Three days after treatment, cells were harvested and counted using a Luna-II Automated Cell Counter (Labtech). Annexin V and Vybrant FAM caspase-3/-7 assays were also used to assess apoptosis. In pooled samples, CCT241736 showed induction of apoptosis as measured by annexin V and caspase-3/-7 assays (Figure 5A; supplemental Figure 5A-M) and a consistent reduction of cell viability independently of FLT3, IDH1/2, or NPM1 status (Figure 5B). Detailed analysis showed that in 6 samples (AML-2, AML-3, AML-6, AML-7, AML-12, AML-13), CCT241736 induced apoptosis as measured by annexin V and caspase-3/-7 assays and consistently reduced cell viability independently of FLT3, IDH1/2, or NPM1 status. Six additional samples (AML-1, AML-4, AML-8, AML-9, AML-10, AML-11) responded to CCT241736 at higher concentrations (3 μM), whereas only 1 sample (AML-5 with FLT3 WT and NPM1) did not respond (supplemental Figure 5A-M). To investigate the cause of cell death, we determined the effect of CCT241736 (1 μM for 4 hours) on P-STAT5 and Aurora pathways in 2 samples of CCT241736-responsive primary AML samples (1 FLT3 WT [AML-2] and 1 FLT3-ITD [AML-6]; Figure 5C-E). Phospho-STAT5, the downstream marker for FLT3 inhibition, was reduced in both samples (Figure 5C). HH3 phosphorylation, indicative of Aurora inhibition, was reduced by ∼25% to 35% in both samples (Figure 5D). Immunoblots are presented in Figure 5E. These data confirm that the potency of CCT241736 in primary AML samples and in in vivo experiments is a result of the inhibition of both FLT3 and Aurora.
Characteristics of primary AML samples from quizartinib-sensitive and quizartinib-relapsed (bold) patients
| Sample | Cytogenetics | Cytogenetic risk group | NPM1 mutation | FLT3 mutation | RUNX1 mutation |
|---|---|---|---|---|---|
| AML-7 | Normal | Intermediate | Mutant | ITD | Not tested |
| AML-7 | Normal | Intermediate | Mutant | ITD | WT |
| AML-14 | t(6;9)(p23;q34.1) | Intermediate | WT | ITD | Not tested |
| AML-14 | t(6;9)(p23;q34.1) | Intermediate | WT | ITD, D835Y* D835del† | Not tested |
| AML-15 | +13 | Intermediate | WT | ITD | Not tested |
| AML-15 | +13 | Intermediate | WT | ITD D835_M837 delinsValLeu‡ | D93fs§ L110Q|| |
| Sample | Cytogenetics | Cytogenetic risk group | NPM1 mutation | FLT3 mutation | RUNX1 mutation |
|---|---|---|---|---|---|
| AML-7 | Normal | Intermediate | Mutant | ITD | Not tested |
| AML-7 | Normal | Intermediate | Mutant | ITD | WT |
| AML-14 | t(6;9)(p23;q34.1) | Intermediate | WT | ITD | Not tested |
| AML-14 | t(6;9)(p23;q34.1) | Intermediate | WT | ITD, D835Y* D835del† | Not tested |
| AML-15 | +13 | Intermediate | WT | ITD | Not tested |
| AML-15 | +13 | Intermediate | WT | ITD D835_M837 delinsValLeu‡ | D93fs§ L110Q|| |
c.2503G>T.
c.2502_2505delinsC.
c.2504_2509delinsTCT.
c.273_277dupCACCG.
c.328A>C.
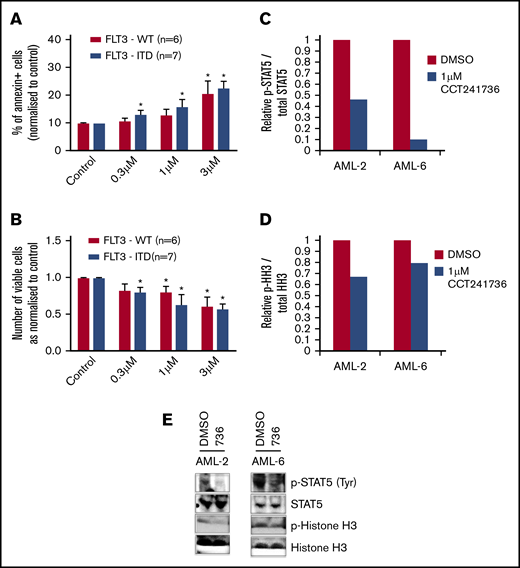
The effect of CCT241736 on cell viability and induction of apoptosis and biomarker modulation of primary AML cells. (A-B) Primary cells from AML samples with FLT3 WT and FLT3-ITD were cocultured with MS-5 and treated with either DMSO control or CCT241736 at indicated doses. (A) Three days after treatment, cells were harvested, washed, and stained with annexin-V/DAPI. (B) Viable cells were counted using a Luna-II Automated Cell Counter. (C-E) Primary AML samples AML-2 (FLT3 WT) and AML-6 (FLT3-ITD) were treated with 1 µM CCT241736 and DMSO for 4 hours, the relative levels of P-STAT5 vs total STAT5 (C), and P-HH3 vs total HH3 (D) were analyzed by immunoblotting using specific antibodies (E). All experiments were performed in triplicate, and results were shown as mean ± SEM. *P < .01 compared with control.
The effect of CCT241736 on cell viability and induction of apoptosis and biomarker modulation of primary AML cells. (A-B) Primary cells from AML samples with FLT3 WT and FLT3-ITD were cocultured with MS-5 and treated with either DMSO control or CCT241736 at indicated doses. (A) Three days after treatment, cells were harvested, washed, and stained with annexin-V/DAPI. (B) Viable cells were counted using a Luna-II Automated Cell Counter. (C-E) Primary AML samples AML-2 (FLT3 WT) and AML-6 (FLT3-ITD) were treated with 1 µM CCT241736 and DMSO for 4 hours, the relative levels of P-STAT5 vs total STAT5 (C), and P-HH3 vs total HH3 (D) were analyzed by immunoblotting using specific antibodies (E). All experiments were performed in triplicate, and results were shown as mean ± SEM. *P < .01 compared with control.
We expanded this investigation by using samples from 3 quizartinib-relapsed patients (AML-7, AML-14, and AML-15; Table 2 [bold]). Further characterization of these samples identified FLT3-TKD mutations in a sample from AML-14 (D835Y and D835del) and in a sample from AML-15 (D835del and M837del). Additional mutations of RUNX1 gene were found in AML-15 (D93fs and L110Q; Table 2).26 None of the above gene alterations were identified in quizartinib-resistant sample from AML-7. We tested cell viability and apoptosis induction of quizartinib and CCT241736 in these samples. Quizartinib showed no effect on any of these parameters, whereas CCT241736 clearly showed a reduction of viable cells (Figure 6) and induction of apoptosis as measured by the induction of caspase-3/-7 activity and annexin V staining (supplemental Figure 6). These results suggest that dual inhibition of FLT3 and Aurora kinases by CCT241736 sensitizes AML cells with various quizartinib-resistant mutations to death.
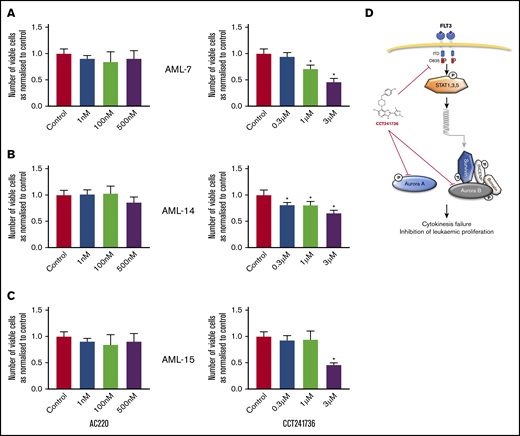
The effect of AC220 and CCT241736 on cell viability of quizartinib-resistant primary AML cells. (A-C) Primary AML cells were collected from 3 patients with quizartinib-resistant AML (AML-7, AML-14, and AML-15). The AML cells were collected after clinical quizartinib resistance had emerged at the point of relapse. Cells were cocultured in triplicate with irradiated MS-5 at a density of 1.2 million cells per milliliter and were treated with either DMSO control, AC220 (left graphs), or CCT241736 (right graphs) at the indicated doses. Three days after treatment, cells were harvested, and viable cells were counted using a Luna-II Automated Cell Counter. All experiments were done in triplicate and results were shown as mean ± SEM (*P < .05 compared with control). (D) Explanatory model: activated FLT3 induces the transcription of several downstream genes, including the Aurora B partner survivin, via STAT1, -3, and -5 proteins. Aurora A plays a role in centrosome maturation and spindle formation, whereas Aurora B regulates the spindle checkpoint in kinetochore microtubule attachment and cytokinesis. CCT241736 inhibits Aurora kinases and the FLT3 pathway in multiple steps. This inhibition leads to cytokinesis failure and inhibition of leukemic proliferation.
The effect of AC220 and CCT241736 on cell viability of quizartinib-resistant primary AML cells. (A-C) Primary AML cells were collected from 3 patients with quizartinib-resistant AML (AML-7, AML-14, and AML-15). The AML cells were collected after clinical quizartinib resistance had emerged at the point of relapse. Cells were cocultured in triplicate with irradiated MS-5 at a density of 1.2 million cells per milliliter and were treated with either DMSO control, AC220 (left graphs), or CCT241736 (right graphs) at the indicated doses. Three days after treatment, cells were harvested, and viable cells were counted using a Luna-II Automated Cell Counter. All experiments were done in triplicate and results were shown as mean ± SEM (*P < .05 compared with control). (D) Explanatory model: activated FLT3 induces the transcription of several downstream genes, including the Aurora B partner survivin, via STAT1, -3, and -5 proteins. Aurora A plays a role in centrosome maturation and spindle formation, whereas Aurora B regulates the spindle checkpoint in kinetochore microtubule attachment and cytokinesis. CCT241736 inhibits Aurora kinases and the FLT3 pathway in multiple steps. This inhibition leads to cytokinesis failure and inhibition of leukemic proliferation.
Discussion
Several small molecule kinase inhibitors with FLT3 activity have been evaluated in relapsed or refractory AML patients in clinical trials. These include lestaurtinib (CEP 701), midostaurin (PKC-412), sorafenib (BAY-43-9006), sunitinib, (SU-11248), and tandutinib (MLN518).27-31 Second-generation inhibitors have now been developed that show greater selectivity and low nanomolar target inhibition; such agents include KW-2449 and quizartinib (AC220).32,33 Although these inhibitors (as monotherapies) have demonstrated moderate efficacy, the emergence of resistance and relapse has been challenging.
Several resistance mechanisms have been identified. Primary resistance involves reduced potency as well as adverse effects on bone marrow leukemia cells and circulating blasts; for example, quizartinib induces apoptosis in the circulating blasts but promotes differentiation of bone marrow blasts.4,34 Secondary resistance also involves regaining sensitivity to FL whose expression is reported to increase immediately after chemotherapy.4,9 In addition, it has been reported that patients who relapse after FLT3 therapy develop point mutations around the adenosine triphosphate–binding pocket or a gatekeeper mutation (D835Y, D835V, D835F, or F691L), thus blocking inhibitor binding.7,35,36 Several FLT3 inhibitors are now being clinically investigated in combination with other agents in an attempt to prevent the occurrence of resistance.37,38
As critical mitotic kinases, Aurora A and Aurora B have been targeted with numerous small molecules. For FLT3-ITD+ AML, several Aurora kinase inhibitors have demonstrated efficacy in early-phase clinical trials.11 In this study, we aimed to characterize CCT241736 as a dual FLT3-Aurora inhibitor both in vitro and in vivo, to determine its mechanism of action, and to evaluate its ability to overcome resistance to selective FLT3 inhibitors.
CCT241736 is a potent dual inhibitor of FLT3 and Aurora kinases with few off-target kinase activities across the kinome, namely JAK2 and Axl.15 JAK2 signaling has been associated with the maintenance of AML stem cells and thus may represent a target in AML. The JAK2 inhibitory activity of CCT241736 may act to prevent the occurrence of resistance.39 Phosphorylation of Axl has been shown to be upregulated after exposure to the FLT3 inhibitor AC220 in both an FLT3-ITD+ AML cell line (MV4-11) and in primary blasts. The Axl inhibitory activity of CCT241736 thus may also act to prevent the occurrence of resistance.40 The PK profile of CCT241736 in mice and rats revealed a highly orally bioavailable compound with low clearance and a moderate volume of distribution.15
In cellular assays, CCT241736 potently inhibited the proliferation of cell lines with both mutated and WT FLT3. This inhibition was associated with the induction of apoptosis as measured by PARP cleavage as well as downregulation of survivin. The cell cycle profile of CCT241736, with temporary G1-S and G2-M arrest, is differentiated from selective FLT3 inhibition and pan-Aurora inhibition and phenocopies cells treated simultaneously with an FLT3 inhibitor (MLN518) and a pan-Aurora inhibitor (PHA-739358). These results together provide an explanatory model for understanding the unique mechanism of action of CCT241736 targeting of AML (Figure 6D).
On the basis of its in vitro pharmacologic profile, we carried out human tumor xenograft experiments in athymic mice to evaluate the efficacy of CCT241736 in vivo. CCT241736 demonstrated dose-dependent reduction of tumor growth in a MOLM-13 subcutaneous human tumor xenograft model with PD modulation of biomarkers, which indicated both FLT3 and Aurora kinase inhibition. Importantly, we have also shown (by immunofluorescence microscopy of tumor sections) that CCT241736 induces abnormal mitosis in MV4-11 tumor cells, a characteristic phenotype of Aurora inhibition. These data suggest that the CCT241736-induced Aurora inhibitory effects also contribute to the observed in vivo efficacy in these models.
To test the hypothesis that dual FLT3-Aurora inhibition could overcome resistance to selective FLT3 inhibition, we have used MOLM-13-RES cells (pD835Y), generated in our laboratory in the presence of increasing concentrations of the selective FLT3 inhibitor MLN518.17 Additional studies showed that CCT241736 retained efficacy against MOLM-13-RES cells grown as subcutaneous xenografts with biomarker modulation compared with MLN518. Long-term treatment of AML xenografts with CCT241736 (over 4 months) failed to show evidence of loss of responsiveness, which builds confidence that the dual pharmacologic profile of this compound (Aurora and FLT3 inhibition) abrogates or significantly delays the development of resistance.
The potency of CCT241736 was also confirmed in other clinically relevant secondary FLT3-TKD mutations in vitro in a panel of mouse BaF3 AML cells that expressed different FLT3 mutants.7 Importantly, we have investigated the in vivo efficacy of CCT241736 in a systemic model by using cells that express FLT3 with a mutated gatekeeper residue (F691) that has been identified in AC220-treated patients who have relapsed. It was shown that CCT241736 inhibited tumor burden by 42%, confirming the significant benefit of CCT241736 treatment in reducing tumor growth in this model when compared with AC220.
Patient samples with FLT3 WT, FLT3-ITD, and WT or mutated NPM1 and IDH1/2 genes were sensitive to CCT241736 with biomarker modulation, demonstrating that CCT241736 is a potent inhibitor of FLT3 and Aurora kinases in primary patient cells. More importantly, sensitization to death of primary cell samples from quizartinib-relapsed AML patients by CCT241736 offers the opportunity to progress a new therapeutic strategy in this particularly aggressive subtype of AML. Overall, the unique kinase inhibitory profile of CCT241736, with inhibitory activity toward both FLT3 and Aurora kinases, may yield an agent that has persistent clinical activity against AML with a low incidence of acquired resistance.
Acknowledgments
The authors acknowledge C. Smith from the University of California, San Francisco, for providing the BaF3-mutant cells expressing FLT3-TKD mutants.
This work (and all authors from the Cancer Research UK Cancer Therapeutics Unit) were supported by core funding from Cancer Research UK (grant nos. C309/A8274 and C309/A11566; and grant no. C1178/A10294 [A.S.M.]), by the National Institute for Health Research Royal Marsden/Institute of Cancer Research Biomedical Research Centre, and by a New Investigator Scholarship from the Haematology Society of Australia and New Zealand (A.S.M.). A.D.J.P. was supported by Cancer Research UK (programme grant no. C1178/A10294). S.L. was supported by the Breast Cancer Now Centre.
Authorship
Contribution: A.S.M. and S.L. designed the study; A.S.M. performed experiments, analyzed data, and wrote the manuscript; A.F. and G.W.Y.M. performed experiments, analyzed data, and helped write parts of the manuscript; G.B., A.H., M.V., A.d.H.B., C.P.R.X., and S.A.E. performed experiments and analyzed data; V.B. synthesized and provided the appropriate compounds; A.D.J.P., F.I.R., J.B., and R.C. discussed and analyzed data; R.S. provided primary samples and analyzed data; F.M.-M. and D.C.T. designed and performed the assays in primary AML samples, analyzed data, and helped write parts of the manuscript; and S.L. directed the project, secured funding, and wrote the manuscript.
Conflict-of-interest disclosure: All authors are employees of The Institute of Cancer Research, which has a commercial interest in drug development programs (www.icr.ac.uk). Please note that all authors who are or have been employed by The Institute of Cancer Research are subject to a Rewards to Inventors Scheme which may reward contributors to a program that is subsequently licensed. J.B. is a stockholder in Azeria Therapeutics and NeoPhore Ltd.
The current affiliation for A.S.M. is Child Health Research Centre, The University of Queensland and Children’s Health Queensland Hospital and Health Service Centre for Children’s Health Research, South Brisbane, Australia.
The current affiliation for A.F. is Lahore University of Management Sciences, Lahore, Pakistan.
The current affiliation for C.P.R.X. is Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Porto, Portugal.
The current affiliation for J.B. is Azeria Therapeutics Ltd, Cambridge, United Kingdom.
S.A.E. is retired from The Institute of Cancer Research, London, United Kingdom.
A.D.J.P. is retired from The Institute of Cancer Resarch and The Royal Marsden Hospital, Sutton, United Kingdom.
Correspondence: Spiros Linardopoulos, The Institute of Cancer Research, 123 Old Brompton Rd, London SW7 3RP, United Kingdom; e-mail: spiros.linardopoulos@icr.ac.uk.

