

Key Points
Lentiviral-mediated ADA2 gene transfer corrects the enzymatic defect in cells of patients with DADA2.
ADA2 reconstitution in patients’ macrophages prevents excessive inflammation.

Abstract
Adenosine deaminase 2 deficiency (DADA2) is a rare inherited disorder that is caused by autosomal recessive mutations in the ADA2 gene. Clinical manifestations include early-onset lacunar strokes, vasculitis/vasculopathy, systemic inflammation, immunodeficiency, and hematologic defects. Anti–tumor necrosis factor therapy reduces strokes and systemic inflammation. Allogeneic hematopoietic stem/progenitor cell (HSPC) transplantation can ameliorate most disease manifestations, but patients are at risk for complications. Autologous HSPC gene therapy may be an alternative curative option for patients with DADA2. We designed a lentiviral vector encoding ADA2 (LV-ADA2) to genetically correct HSPCs. Lentiviral transduction allowed efficient delivery of the functional ADA2 enzyme into HSPCs from healthy donors. Supranormal ADA2 expression in human and mouse HSPCs did not affect their multipotency and engraftment potential in vivo. The LV-ADA2 induced stable ADA2 expression and corrected the enzymatic defect in HSPCs derived from DADA2 patients. Patients’ HSPCs re-expressing ADA2 retained their potential to differentiate into erythroid and myeloid cells. Delivery of ADA2 enzymatic activity in patients’ macrophages led to a complete rescue of the exaggerated inflammatory cytokine production. Our data indicate that HSPCs ectopically expressing ADA2 retain their multipotent differentiation ability, leading to functional correction of macrophage defects. Altogether, these findings support the implementation of HSPC gene therapy for DADA2.
Introduction
Adenosine deaminase 2 deficiency (DADA2) is a rare inherited disorder that is caused by autosomal recessive mutations in the ADA2 gene.1,2 Disease onset is usually in childhood, and a significant proportion of patients die early in life.1-4 DADA2 patients present with a variable clinical phenotype, including cutaneous and cerebral vasculopathy, ranging from livedo reticularis and polyarteritis nodosa to life-threatening intracranial hemorrhages and lacunar strokes. Systemic inflammation similarly affects the kidney, liver, and gastrointestinal tract. The clinical spectrum also includes hematological and immunological manifestations, such as cytopenias (neutropenia, autoimmune hemolytic anemia, thrombocytopenia, severe pure red cell aplasia),1,3,5 mild immunodeficiency, hypogammaglobulinemia, low switched memory B cells, and low immunoglobulin M serum levels.6,7 Features that mimic autoimmune lymphoproliferative syndrome can also occur in DADA2.8–10
ADA2 shares partial structural homology with ADA1, a well-known enzyme catalyzing adenosine and deoxyadenosine deamination into inosine and deoxyinosine, respectively. Although ADA1 and ADA2 share adenosine deaminase activity, they differ substantially. ADA1 is a monomeric intracellular protein that is expressed in most cell types, but significant amounts can also be found in plasma. ADA2 is a secreted homodimeric protein that is produced by myeloid cells (monocytes, macrophages).11 The affinity of ADA2 for adenosine substrate is much weaker than that of ADA1.12 Based on these observations, ADA1 should represent the primary regulator of extracellular adenosine levels, whereas ADA2 should act locally at the site of inflammation, where adenosine levels are much higher. The most striking evidence that the 2 ADA isoforms have nonredundant biological functions derives from the observation that ADA1 and ADA2 deficiencies manifest with distinct pathological features. Patients with ADA1 deficiency exhibit a severe combined immunodeficiency with no evidence of vasculopathy, systemic inflammation, or hematological defects typical of DADA2.
How ADA2 regulates the immune system is still enigmatic. Increased ADA2 levels were found in fluids from patients with infections (Mycobacterium tuberculosis13 and HIV-114) and chronic inflammatory conditions (systemic lupus erythematosus,15 rheumatoid arthritis,16 macrophage activation syndrome,17 Crohn’s disease,18 chronic active hepatitis19). Defective B-cell and T follicular helper cell responses have been reported.20 Proinflammatory cytokines (interleukin-1β [IL-1β], tumor necrosis factor [TNF]) were elevated in the skin and brain biopsies of patients.1 Loss of ADA2 in patients is associated with increased production of proinflammatory cytokines from M1 macrophages and poor differentiation of M2 macrophages.1 DADA2 is associated with enhanced neutrophil extracellular trap formation.21 Marked upregulation of neutrophil-expressed genes and an interferon (IFN) signature in patients’ blood leukocytes were also reported.22,23 All of these observations suggest that DADA2 pathophysiology may derive from a dysregulated activation of the myeloid cell compartment, causing endothelial cell activation and vascular disease.
Because inflammation is one of the overarching features of DADA2, current medical management is based on standard immunosuppression. Anti-TNF therapy reduced vasculitis and prevented strokes.24,25 Allogeneic hematopoietic stem cell transplantation (HSCT) has shown promise in small cohorts of DADA2 patients with the hematological phenotype.8,26,27 However, HLA-matched donors are not always available, and the risk of morbidity related to HSCT should not be underestimated in DADA2 patients with vasculitis, systemic inflammation, and immunodeficiency.27 Therefore, the development of alternative therapeutic approaches for DADA2 is urgently needed. It is reasonable to assume that strategies based on genetic correction and engraftment of autologous hematopoietic stem/progenitor cells (HSPCs) may provide a definitive cure for DADA2. This study evaluated whether a lentiviral vector encoding ADA2 (LV-ADA2) can restore enzymatic activity in patients’ HSPCs and correct macrophage inflammatory activation.
Methods
Patients
Patients 1 and 2 (twins) carry a heterozygous substitution at c.563T>C (p.Leu188Pro) and exon 7 deletion (IVS6_IVS7del). Patient 3 carries a homozygous missense mutation c.1367A>G (p.Tyr456Cys). Patient 4 carries a homozygous substitution c.139G>A (p.Gly47Arg). Patient 5 carries a deletion in exon 2, c.144del (p.Arg49Glyfs), and the nucleotide substitution c.1085G>A (p.Trp362Ter). Patients 6 and 7 (siblings) carry the missense mutations c.140G>T (p.Gly47Val) in exon 2 and c.1435T>C (p.Ser479Pro) in exon 9. Table 1 describes patient characteristics.
Characteristics of the patients included in the study
| ID | Sex | Age, y | Mutations | Phenotype | IS therapy* | G-CSF |
|---|---|---|---|---|---|---|
| 1 | F | 23 | p.Leu188Pro/IVS6_IVS7del* | 2 strokes (2-3 y), hypogammaglobulinemia (3 y), recurrent infections (since 13 y), livedo reticularis (13 y), neutropenia (14 y), TLGL (21 y) | Etanercept, PD | Yes |
| 2 | F | 23 | p.Leu188Pro/IVS6_IVS7del* | 2 strokes (8-14 y), neutropenia (8 y), hypogammaglobulinemia (8 y), livedo reticularis (10 y), hypothyroidism (22 y) | Etanercept | Yes |
| 3 | F | 31 | p.Tyr456Cys/p.Tyr456Cys | Hodgkin lymphoma (28 y), severe prolonged neutropenia after chemotherapy (30 y) | MPD | No |
| 4 | F | 18 | p.Gly47Arg/p.Gly47Arg | Livedo reticularis (early infancy), recurrent giardiasis (3-4 y), PAN (5 y), hemorrhagic stroke (10 y), intracranial aneurysm rupture (17 y), hypertension, systemic inflammation | Etanercept | No |
| 5 | F | 63 | p.Arg49Glyfs*/p.Trp362Ter | Oral candidiasis (1 mo), RRI (infancy), hypogammaglobulinemia (14 y), neutropenia (20 y), recurrent pneumonias (45-50 y), TLGL (61 y) | Etanercept, rituximab | Yes |
| 6 | F | 24 | p.Gly47Val/p.Ser479Pro | Livedo (7 y), neurological symptoms (8 y), PAN and hypertension (9 y), pericarditis (10 y), peripheral neuropathy (12 y), hypo-IgM (24 y), systemic inflammation | Etanercept | No |
| 7 | M | 25 | p.Gly47Val/p.Ser479Pro | Neurological symptoms (2 y), hypertension (10 y), PAN (11 y), stroke (11 y), hypogammaglobulinemia (25 y), systemic inflammation | Etanercept | No |
| ID | Sex | Age, y | Mutations | Phenotype | IS therapy* | G-CSF |
|---|---|---|---|---|---|---|
| 1 | F | 23 | p.Leu188Pro/IVS6_IVS7del* | 2 strokes (2-3 y), hypogammaglobulinemia (3 y), recurrent infections (since 13 y), livedo reticularis (13 y), neutropenia (14 y), TLGL (21 y) | Etanercept, PD | Yes |
| 2 | F | 23 | p.Leu188Pro/IVS6_IVS7del* | 2 strokes (8-14 y), neutropenia (8 y), hypogammaglobulinemia (8 y), livedo reticularis (10 y), hypothyroidism (22 y) | Etanercept | Yes |
| 3 | F | 31 | p.Tyr456Cys/p.Tyr456Cys | Hodgkin lymphoma (28 y), severe prolonged neutropenia after chemotherapy (30 y) | MPD | No |
| 4 | F | 18 | p.Gly47Arg/p.Gly47Arg | Livedo reticularis (early infancy), recurrent giardiasis (3-4 y), PAN (5 y), hemorrhagic stroke (10 y), intracranial aneurysm rupture (17 y), hypertension, systemic inflammation | Etanercept | No |
| 5 | F | 63 | p.Arg49Glyfs*/p.Trp362Ter | Oral candidiasis (1 mo), RRI (infancy), hypogammaglobulinemia (14 y), neutropenia (20 y), recurrent pneumonias (45-50 y), TLGL (61 y) | Etanercept, rituximab | Yes |
| 6 | F | 24 | p.Gly47Val/p.Ser479Pro | Livedo (7 y), neurological symptoms (8 y), PAN and hypertension (9 y), pericarditis (10 y), peripheral neuropathy (12 y), hypo-IgM (24 y), systemic inflammation | Etanercept | No |
| 7 | M | 25 | p.Gly47Val/p.Ser479Pro | Neurological symptoms (2 y), hypertension (10 y), PAN (11 y), stroke (11 y), hypogammaglobulinemia (25 y), systemic inflammation | Etanercept | No |
F, female; G-CSF, granulocyte colony-stimulating factor; IgM, immunoglobulin M; IS, immunosuppressive; M, male; MPD, methylprednisolone; PAN, polyarteritis nodosa; PD, prednisone; RRI, recurrent respiratory infections; TLGL, T large granular lymphocytes.
Ongoing treatment at the time of blood collection.
Study approval
All clinical investigations were conducted according to Declaration of Helsinki principles. Patients, parents, and healthy controls gave informed consent following a standard ethical procedure (clinical protocol number Tiget 06) approved by the Ethics Committees of IRCCS Ospedale San Raffaele (Milan) and IRCCS Ospedale Giannina Gaslini (Genoa). Bone marrow (BM) and peripheral blood were collected from DADA2 patients during diagnostic or therapeutic procedures. The relevant institutional review boards approved the research.
Mouse studies were conducted according to protocols approved by the IRCCS San Raffaele Scientific Institute and Institutional Animal Care and Use Committee (IACUC #997), adhering to the Italian Ministry of Health guidelines for the use and care of experimental animals. All efforts were made to minimize the number, pain, and distress of mice during and after experimental procedures.
Mice
NOD.Cg-KitW-41JPrkdcscidIl2rgtm1Wjl/WaskJ (NSGW41; stock #026497) mice were purchased from The Jackson Laboratory. C57BL/6-Ly5.1 and C57BL/6N mice were purchased from Charles River (Calco, Italy). Mice were maintained in specific pathogen–free conditions at the IRCCS San Raffaele Scientific Institute SPF Animal Facility.
Lentiviral transduction of human CD34+ cells
BM CD34+ cells isolated from healthy donors (HDs) were purchased from Lonza. Patients’ CD34+ cells were purified from BM aspirate using a CD34 MicroBead Kit (Miltenyi Biotec). Cell purity was >97%. Cells (1 × 106 cells per milliliter, 0.4-1.1 × 105 cells per well), resuspended in serum-free Cellgro medium containing human stem cell factor (SCF) (300 ng/mL), thrombopoietin (TPO) (100 ng/mL), fms related receptor tyrosine kinase 3 ligand (FLT3L) (300 ng/mL), and IL-3 (60 ng/mL; all from PeproTech), were incubated in RetroNectin-coated wells for 22 hours. 16,16-Dimethyl-prostaglandin E2 (10 μM) was added 2 hours before lentiviral vector (LV) transduction at the indicated multiplicity of infection (MOI). After a 16-hour transduction, cells were collected, washed, and cultured in IMDM supplemented with fetal bovine serum (FBS), 1% l-glutamine, 1% penicillin/streptomycin, SCF (100 ng/mL), TPO (100 ng/mL), Flt3L (100 ng/mL), and IL-3 (20 ng/mL) for 14 days.
Colony-forming assay
Purified CD34+ cells were resuspended in complete human MethoCult medium and plated at 1000 cells per plate in 35-mm plates in duplicate per condition. Fifteen days later, erythroid burst-forming units (BFU-E), granulocyte-macrophage colony-forming units (CFU-GM), and granulocyte, erythroid, macrophage, megakaryocyte colony-forming units (CFU-GEMM) were assessed for number and morphology by light microscopy.
Macrophage transduction and polarization
CD14+ monocytes were purified from peripheral blood mononuclear cells using CD14 MicroBeads (Miltenyi Biotec), according to the manufacturer’s instructions. Monocytes were plated in RPMI 1640 supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine in the presence of human recombinant macrophage colony-stimulating factor (M-CSF) (10 ng/mL). For transduction, monocytes were incubated for 2 hours with the accessory viral protein vpl-VPX, followed by overnight transduction at the MOI of 10. Viral-containing supernatant was removed, and cells were incubated with M-CSF (10 ng/mL) in complete medium for 4 days. For M1 polarization, macrophages were treated with Escherichia coli lipopolysaccharide (LPS; 1 µg/mL) and human recombinant IFN-γ (20 ng/mL) for 48 hours.
U937 cells were cultured in RPMI 1640 supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine and differentiated into macrophages by incubation with 12-O-tetradecanoylphorbol-13-acetate (20 ng/mL) for 48 hours. U937 macrophages were cultured in complete medium devoid of 2-O-tetradecanoylphorbol-13-acetate for 1 day and then polarized into M1-like macrophages with LPS (50 ng/mL) and IFN-γ (10 ng/mL) for 48 hours.
Statistical methods
GraphPad Prism version 9 (GraphPad Software) was used to prepare graphics and perform statistical analyses. The Mann-Whitney U test was used to compare the distribution of a numerical variable among 2 independent groups. The rank-based nonparametric Kruskal-Wallis test was used to determine statistically significant differences between ≥2, and the nonparametric Wilcoxon signed-rank test was used to compare 2 matched samples. P values ≤ .05 were considered statistically significant.
Results
LV-mediated transduction directed the efficient delivery of ADA2 expression and enzymatic activity in human CD34+ cells without evidence of toxicity
We explored the ADA2 expression profile in various subsets of peripheral blood mononuclear cells and BM progenitors. ADA2 messenger RNA and protein were expressed more strongly in CD14+ monocytes and, at lower levels, in T (CD4+ and CD8+) cells, B (CD19+) cells, natural killer (NK; CD56+) cells, and BM CD34+ HSPCs (supplemental Figure 1).
Based on the broad expression profile in immune cells, we generated an LV-ADA2 in which the expression of human ADA2 was driven by the ubiquitous human phosphoglycerate kinase (PGK) promoter (Figure 1A). The PGK promoter allows stable expression of therapeutic proteins in myeloid cells28 and has been used at San Raffaele Telethon Institute for Gene Therapy to transduce CD34+ cells in >40 patients affected by metabolic disorders with a favorable risk-benefit profile for up to 8 years after gene therapy.29 A PGK.GFP LV was used as a negative control vector (LV-GFP). Human CD34+ HSPCs isolated from the BM of 3 independent HDs were transduced with LV-ADA2 or LV-GFP at an MOI of 10, 50, and 100 in the presence of prostaglandin E2 as a transduction enhancer.30,31 The average transduction efficiency, evaluated on individual vector-positive CFU-GM and BFU-E, was ≥75% in all conditions (Figure 1B). The median vector copy number (VCN) ranged from 1.82 to 2.69 and from 1.58 to 3.70 in the CFU-GM and BFU-E colonies, respectively (Figure 1C). The viability of CD34+ cells pre- and post–LV-ADA2 exposure was comparable to cells cultured in the absence (untransduced; UT) or presence of LV-GFP (Figure 1D). LV-ADA2 transduction did not affect the clonogenic potential of CD34+ cells at each vector dose (Figure 1E), indicating a lack of toxicity, even at a high VCN.
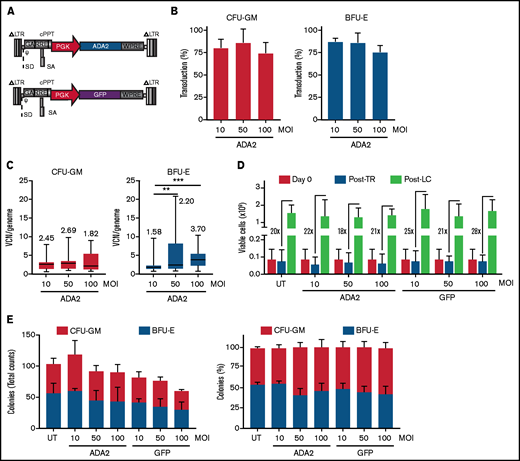
Efficient LV-mediated ADA2 transfer in HDs’ HSPCs. (A) Schematic representation of LV-ADA2 and LV-GFP used in the study. Percentage of transduction (B) and VCN per genome (C) were evaluated in colonies derived from CD34+ HSPCs from 3 independent HDs transduced with LV-ADA2 at increasing vector concentrations (MOI of 10, 50, and 100). The numbers in panel C indicate the median VCN for each condition. (D) The viability of HDs’ CD34+ HSPCs in BM was examined before prestimulation (day 0), postovernight transduction (Post-TR) with LV-ADA2 and LV-GFP, and after in vitro expansion for 14 days (Post-LC) in the presence of support cytokines. Numbers indicate the expansion rate between in vitro expanded cells (Post-LC) and cells recovered after transduction (Post-TR). (E) Total numbers and percentages of single colonies derived from myeloid (CFU-GM) and erythroid (BFU-E) progenitors in UT and ADA2- or GFP-transduced conditions. Data in (B,D-E) are mean ± standard deviation; data in (C) are shown as box-and-whisker plots. **P < .01, ***P < .001; Mann-Whitney U test.
Efficient LV-mediated ADA2 transfer in HDs’ HSPCs. (A) Schematic representation of LV-ADA2 and LV-GFP used in the study. Percentage of transduction (B) and VCN per genome (C) were evaluated in colonies derived from CD34+ HSPCs from 3 independent HDs transduced with LV-ADA2 at increasing vector concentrations (MOI of 10, 50, and 100). The numbers in panel C indicate the median VCN for each condition. (D) The viability of HDs’ CD34+ HSPCs in BM was examined before prestimulation (day 0), postovernight transduction (Post-TR) with LV-ADA2 and LV-GFP, and after in vitro expansion for 14 days (Post-LC) in the presence of support cytokines. Numbers indicate the expansion rate between in vitro expanded cells (Post-LC) and cells recovered after transduction (Post-TR). (E) Total numbers and percentages of single colonies derived from myeloid (CFU-GM) and erythroid (BFU-E) progenitors in UT and ADA2- or GFP-transduced conditions. Data in (B,D-E) are mean ± standard deviation; data in (C) are shown as box-and-whisker plots. **P < .01, ***P < .001; Mann-Whitney U test.
Next, we examined ADA2 expression and activity in HD CD34-derived myeloid cells at 14 days posttransduction. An increase in the protein and transcript ADA2 levels was observed in LV-ADA2–transduced, but not in LV-GFP–transduced, cells (Figure 2A-B). Vector-derived ADA2 protein exhibiting a proper enzymatic activity was also secreted from ADA2-transduced cells in a dose-dependent manner (Figure 2C-D). Moreover, proinflammatory M1 macrophages differentiated from LV-ADA2–transduced HD CD34+ cells released TNF and IL-6 at similar levels as did LV-GFP–transduced and UT macrophages (supplemental Figure 2).

Robust ADA2 expression, secretion, and enzymatic activity in CD34-derived cells after LV-ADA2 transduction. ADA2 expression was measured at transcript (A) and protein (B) levels in expanded CD34-derived cells from HDs that were left UT or transduced with the LV-ADA2 and LV-GFP at an MOI of 10, 50, and 100. GAPDH was included as a loading control. Levels of secreted ADA2 are also shown. ADA2 protein (C) and enzymatic activity (D) were measured in cell-free supernatants collected from cells transduced with LV-ADA2 and LV-GFP at an MOI of 10, 50, and 100. Data are mean ± standard deviation. *P < .05.
Robust ADA2 expression, secretion, and enzymatic activity in CD34-derived cells after LV-ADA2 transduction. ADA2 expression was measured at transcript (A) and protein (B) levels in expanded CD34-derived cells from HDs that were left UT or transduced with the LV-ADA2 and LV-GFP at an MOI of 10, 50, and 100. GAPDH was included as a loading control. Levels of secreted ADA2 are also shown. ADA2 protein (C) and enzymatic activity (D) were measured in cell-free supernatants collected from cells transduced with LV-ADA2 and LV-GFP at an MOI of 10, 50, and 100. Data are mean ± standard deviation. *P < .05.
These results indicate that the transduction procedure and LV-derived ADA2 expression are well tolerated and do not influence the capacity of HSPCs to differentiate into myeloid and erythroid cells, as well as that HSPC-derived myeloid cells can mount a proper inflammatory response in vitro.
ADA2 transduction did not impact the multilineage engraftment potential of BM CD34+ cells in vivo
We examined the engrafting and differentiation potential of ADA2-transduced CD34+ cells in vivo. HD CD34+ cells transduced with LV-ADA2 or LV-GFP were transplanted into NSGW41 mice lacking T, B, and NK cells in the absence of preconditioning.32 Vector-positive leukocytes in the peripheral blood were comparable between ADA2 and GFP mice (Figure 3A). The percentage and the absolute number of human CD45+ cells increased over time in both groups (Figure 3B). The absolute counts and the kinetics of the reconstitution of all peripheral blood leukocyte subsets (B, T, myeloid, and NK cells) were similar in the ADA2 and GFP groups (Figure 3C). Moreover, uncommitted hematopoietic stem cells (HSCs), myeloid, lymphoid, and erythroid progenitors, and differentiated cells evaluated in the BM at 20 weeks did not differ between mice receiving ADA2- or GFP-transduced CD34+ cells (Figure 3D), indicating that LV-derived ADA2 expression does not impact the ability of CD34+ cells to engraft, self-renew, or give rise to a multilineage repertoire.
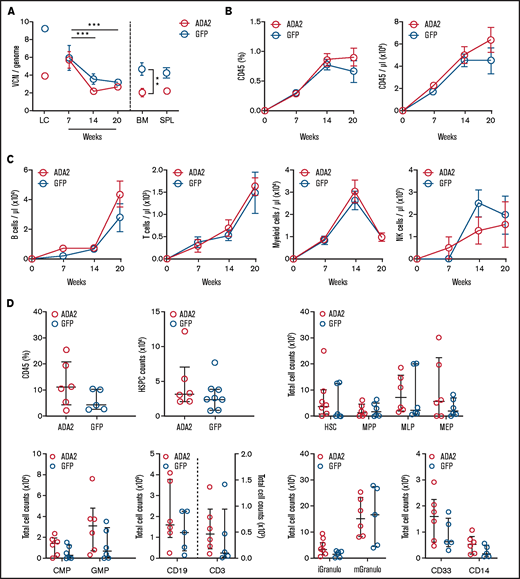
Normal engraftment and multilineage differentiation of ADA2-transduced HDs’ CD34+ cells in NSGW41 mice. (A) The average VCN per genome was evaluated in ADA2- and GFP-transduced HSPCs expanded in vitro (LC), in total peripheral blood leukocytes at 7, 14, and 20 weeks posttransplant, and at 20 weeks in the BM and spleen (SPL) of NSGW41 mice receiving LV-ADA2– or LV-GFP–transduced lineage-negative cells (n = 6 mice per group). Longitudinal analysis of human cell chimerism (CD45+ cells; B) and B, T, myeloid, and NK cells (C) in peripheral blood at 7, 14, and 20 posttransplantation. (D) Flow cytometry analysis showing the total number of HSCs and various progenitor populations in the BM of NSGW41 mice. Data are mean ± standard error. **P < .01, ***P < .001. iGranulo, immature granulocytes; mGranulo, mature granulocytes.
Normal engraftment and multilineage differentiation of ADA2-transduced HDs’ CD34+ cells in NSGW41 mice. (A) The average VCN per genome was evaluated in ADA2- and GFP-transduced HSPCs expanded in vitro (LC), in total peripheral blood leukocytes at 7, 14, and 20 weeks posttransplant, and at 20 weeks in the BM and spleen (SPL) of NSGW41 mice receiving LV-ADA2– or LV-GFP–transduced lineage-negative cells (n = 6 mice per group). Longitudinal analysis of human cell chimerism (CD45+ cells; B) and B, T, myeloid, and NK cells (C) in peripheral blood at 7, 14, and 20 posttransplantation. (D) Flow cytometry analysis showing the total number of HSCs and various progenitor populations in the BM of NSGW41 mice. Data are mean ± standard error. **P < .01, ***P < .001. iGranulo, immature granulocytes; mGranulo, mature granulocytes.
To investigate whether ADA2 overexpression can affect HSPC engraftment and differentiation in a complete null ADA2 background, we generated mouse chimeras, because mice lack an ADA2 ortholog. Lineage-negative cells isolated from the BM of CD45.1 C57BL/6 mice were transduced with LV-ADA2 or LV-GFP and adoptively transferred into lethally irradiated CD45.2 C57BL/6 mice. The median VCN in blood leukocytes was similar between ADA2 and GFP mice and remained stable for up to 19 weeks posttransplant (supplemental Figure 3A). Plasma enzymatic activity in ADA2 chimeras was within the range in plasma from human adult HDs (7.0-25.2 U/L) (supplemental Figure 3B). ADA2 chimeras gained normal weight and did not show any sign of distress (supplemental Figure 3C). Normal tissue architecture and cellularity were observed in the spleen and BM of ADA2 and GFP chimeras (supplemental Figure 3D). The percentage of donor cells, myeloid and lymphoid cell counts in peripheral blood were comparable between ADA2 and GFP chimeras (supplemental Figure 4). We evaluated whether ADA2 overproduction might influence the inflammatory response in vivo. Upon LPS treatment, ADA2 and GFP chimeras produced a similar plasma cytokine profile (supplemental Figure 5).
Altogether, these results indicate that ADA2-expressing human and mouse HSPCs maintain long-term repopulation and multilineage differentiation potential and that ectopic ADA2 overexpression does not exacerbate the inflammatory cytokine response in vivo.
BM of patients with the hematological phenotype of DADA2 exhibited a substantial reduction in HSC and progenitor pools
Despite evidence of hematological alterations, no study has described the phenotypic characteristics of the BM cells of patients with DADA2. Thus, we performed an in-depth study, using multiparametric flow cytometry, of the BM cell composition of adult patients: 2 presenting with severe pancytopenia and BM aplasia requiring allogeneic HSCT (patients 1 and 3) and 1 (patient 2; twin of patient 1) with autoinflammatory manifestations and neutropenia. A cohort of adult HDs was used as a reference dataset for comparative analysis with patients’ BM samples. Compared with HDs, patients’ BM exhibited a reduced number of mature and immature populations belonging to different hematopoietic lineages. The amount of myeloid, B, and erythroid cells was low in all DADA2 patients (Figure 4A-C). Defects in the general hematopoietic output can be attributed to a diminished HSPC number. Indeed, the number of HSCs and committed progenitors of multiple lineages (lymphoid, myeloid, erythroid, and megakaryocyte progenitors) were significantly reduced in patients’ BM compared with HD controls (Figure 4D-E). These immunophenotyping data suggest that DADA2, through cell-intrinsic regulation or extrinsic factors (ie, inflammation, autoreactive antibodies), affects the maintenance of HSCs, giving rise in the long-term to single- or multilineage cytopenia that is typical of patients with hematological and immunological manifestations.

Patients’ BM contains a reduced number of HSCs and progenitors. (A-E) The absolute number of various cell lineages and progenitors was estimated in the BM of 3 patients with DADA2 by multidimensional flow cytometry and compared with a cohort of 12 adult HDs. Data are presented as violin plots. CMP, common myeloid progenitors; DC, dendritic cells; EP, erythroid progenitors. ETP, early T-cell progenitors; GMP, granulocyte-monocyte progenitors; iPMN, immature polymorphonuclear cells; MEP, megakaryocyte-erythroid progenitors; MKp, megakaryocyte progenitors; MLP, multilymphoid progenitors; MPP, multipotent progenitors; PMN, mature polymorphonuclear cells; Pre-BNK, pre-B and NK progenitors; Pro-lympho, pro-lymphocytes. *P < .05, **P < .01.
Patients’ BM contains a reduced number of HSCs and progenitors. (A-E) The absolute number of various cell lineages and progenitors was estimated in the BM of 3 patients with DADA2 by multidimensional flow cytometry and compared with a cohort of 12 adult HDs. Data are presented as violin plots. CMP, common myeloid progenitors; DC, dendritic cells; EP, erythroid progenitors. ETP, early T-cell progenitors; GMP, granulocyte-monocyte progenitors; iPMN, immature polymorphonuclear cells; MEP, megakaryocyte-erythroid progenitors; MKp, megakaryocyte progenitors; MLP, multilymphoid progenitors; MPP, multipotent progenitors; PMN, mature polymorphonuclear cells; Pre-BNK, pre-B and NK progenitors; Pro-lympho, pro-lymphocytes. *P < .05, **P < .01.
LV-mediated ADA2 expression in patients’ CD34+ cells restored enzymatic activity
We investigated the ability of LV-ADA2 to restore ADA2 expression and enzymatic activity in patients’ CD34+ cells. Purified BM CD34+ cells from patients 2 and 3 were cultured in the presence or absence of LV-ADA2 and LV-GFP at an MOI of 10 and 50. CD34+ cells from patients 2 and 3 exhibited a normal capacity to produce CFU-GM, CFU -GEMM, and BFU-E colonies (Figure 5A), suggesting no disruption in their clonogenic capacity following genetic manipulation, despite the overall reduced content. The CFU assay showed a dose-effect relationship between vector dose and transduction efficiency that reached >75% at an MOI of 50 for both vectors; the median VCN per genome was ∼2.3 at an MOI of 50 (Figure 5B-C).

Efficient transduction of patients’ CD34+ cells using the ADA2-LV. (A) Number and percentage of myeloid (CFU-GM), erythroid (BFU-E), and mixed (CFU-GEMM) colonies derived from CD34+ cells from 2 patients and 2 HDs left untreated (UT) or transduced with LV-ADA2 or LV-GFP at an MOI of 10 and 50. (B) Transduction efficiency was calculated as the proportion of vector-positive colonies. (C) Average VCN per genome was evaluated on positive colonies by droplet digital polymerase chain reaction. *P < .05, **P < .01, ***P < .001. PT, patient.
Efficient transduction of patients’ CD34+ cells using the ADA2-LV. (A) Number and percentage of myeloid (CFU-GM), erythroid (BFU-E), and mixed (CFU-GEMM) colonies derived from CD34+ cells from 2 patients and 2 HDs left untreated (UT) or transduced with LV-ADA2 or LV-GFP at an MOI of 10 and 50. (B) Transduction efficiency was calculated as the proportion of vector-positive colonies. (C) Average VCN per genome was evaluated on positive colonies by droplet digital polymerase chain reaction. *P < .05, **P < .01, ***P < .001. PT, patient.
We next measured ADA2 protein expression in UT and transduced CD34+ patient cells maintained in vitro with cytokine support for 14 days. ADA2 was expressed in UT HDs’ cells but was absent in patients’ cells (Figure 6A), indicating that mutations are associated with a complete loss of ADA2 in HSPCs. CD34+ cell transduction with patients' cells LV-ADA2 re-established intracellular ADA2 expression in patients in a dose-dependent manner (Figure 6A). LV-derived ADA2 released from ADA2-transduced cells from patient 3 exhibited a dose-dependent enzymatic activity by increasing vector dose (Figure 6B-C). These findings indicate that LV-ADA2 is an efficient tool to stably deliver ADA2 expression and re-establish enzymatic activity in patients’ CD34+ cells.
![LV-ADA2 restores ADA2 expression and enzymatic activity in patients’ CD34-derived cells. ADA2 expression was evaluated in CD34-derived cells (A) and supernatants (B), from 2 HDs and 2 patients, that were transduced with LV-ADA2 or LV-GFP at an MOI of 10 and 50. β-actin expression was included as a loading control. (C) ADA2 enzymatic activity was quantified in cell-free supernatants from ADA2- and GFP-transduced CD34-derived cells from 1 HD (HD1) and from 1 patient (patient 2 [PT2]).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/16/10.1182_bloodadvances.2020003811/2/m_advancesadv2020003811f6.png?Expires=1765883521&Signature=yywr-VqvBdVitWIit5R1ttN8ZouzK~2SY6OWqDOj7Xko7P1XswAnZmWEBGBxGdr2x3MikxXAkOe1Peafm990tdSRd1ta8lCv7yfbZN9XJy5tfFCvv-iGOn~OurFMKT-vCMWAp2xE-eSMcukpwnt6rcmxsR2IB5zFPk4nP8CQTZtTxgzBb5go3o3cw1h5QILkpB0NXIxfIn0goWDG2NHqnwwUp40hoMLUjLJicp4bQSNFYTsie8d-yoccOhnwCqBqc1q-CV40S0qTjlw87fXki74NqNm59fIja6mUOwAilkfTV7aNKAMNbYa5YVMjPL3SINj6v5HJ-BZms6DQ2kOhKA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
LV-ADA2 restores ADA2 expression and enzymatic activity in patients’ CD34-derived cells. ADA2 expression was evaluated in CD34-derived cells (A) and supernatants (B), from 2 HDs and 2 patients, that were transduced with LV-ADA2 or LV-GFP at an MOI of 10 and 50. β-actin expression was included as a loading control. (C) ADA2 enzymatic activity was quantified in cell-free supernatants from ADA2- and GFP-transduced CD34-derived cells from 1 HD (HD1) and from 1 patient (patient 2 [PT2]).
LV-ADA2 restores ADA2 expression and enzymatic activity in patients’ CD34-derived cells. ADA2 expression was evaluated in CD34-derived cells (A) and supernatants (B), from 2 HDs and 2 patients, that were transduced with LV-ADA2 or LV-GFP at an MOI of 10 and 50. β-actin expression was included as a loading control. (C) ADA2 enzymatic activity was quantified in cell-free supernatants from ADA2- and GFP-transduced CD34-derived cells from 1 HD (HD1) and from 1 patient (patient 2 [PT2]).
ADA2 gene transfer corrected the proinflammatory profile of patients’ macrophages
DADA2 is associated with an increased proinflammatory macrophage profile.1 To study whether LV-derived ADA2 corrects the hyperinflammatory macrophage phenotype, monocyte-derived macrophages from 6 patients were transduced with LV-ADA2 and differentiated into M1 macrophages. ADA2 protein was absent in untreated macrophages from all patients (Figure 7A). Macrophage transduction with LV-ADA2 re-established extracellular ADA2 enzymatic activity (Figure 7B). Although patients’ M1 macrophages showed increased IL-6 and TNF expression despite anti-TNF treatment, LV-mediated ADA2 reconstitution led to a significant reduction in IL-6 expression in all patients, whereas TNF expression decreased in 3 of 5 patients (Figure 7C). IL-6 and TNF release was significantly reduced in patients’ M1 macrophages after ADA2 reconstitution (Figure 7D). Similar results were obtained using M1 macrophages differentiated from ADA2-transduced CD34+ cells from patient 1 (supplemental Figure 6). These data provide the first evidence that LV-mediated restoration of ADA2 corrects the inflammatory macrophage defect in DADA2.

LV-mediated ADA2 correction in patients’ macrophages restored ADA2 enzymatic activity and suppressed cytokine hyperproduction. (A) ADA2 protein expression was assessed in monocyte-derived macrophages from 6 patients with DADA2 and from HDs using immunoblot analysis. β-actin expression was included as a loading control. (B) ADA2 enzymatic activity was measured in cell-free supernatants from patients’ (n = 5) and HDs’ (n = 7) macrophages that were transduced or not with ADA2-LV. IL-6 and TNF expression and release were assessed in UT and ADA2-transduced M1 macrophages from patients and HDs using quantitative reverse transcription polymerase chain reaction (RT-PCR) (C) and enzyme-linked immunosorbent assay (ELISA) (D), respectively. HDs, n = 5; patients, n = 6, with the exception of RT-PCR for TNF expression (n = 5). (E) Immunoblot analysis of ADA2 expression and secretion ADA2+/+ and ADA2−/− U937 macrophages transduced with LV-ADA2 wild-type (ADA2wt) or L188P mutant (ADA2L188P). (F) ADA2 activity was measured in cell-free supernatants from ADA2+/+ and ADA2−/− U937 macrophages transduced with ADA2WT and ADA2L188P. Expression and secretion of IL-6 and TNF were measured in ADA2−/− U937 macrophages expressing ADA2 wild-type or L188P mutant by quantitative RT-PCR (G) and ELISA (H). Data in (F-H) are mean ± standard deviation. *P < .05, **P < .01, ***P < .001. ns, not significant; PT, patient.
LV-mediated ADA2 correction in patients’ macrophages restored ADA2 enzymatic activity and suppressed cytokine hyperproduction. (A) ADA2 protein expression was assessed in monocyte-derived macrophages from 6 patients with DADA2 and from HDs using immunoblot analysis. β-actin expression was included as a loading control. (B) ADA2 enzymatic activity was measured in cell-free supernatants from patients’ (n = 5) and HDs’ (n = 7) macrophages that were transduced or not with ADA2-LV. IL-6 and TNF expression and release were assessed in UT and ADA2-transduced M1 macrophages from patients and HDs using quantitative reverse transcription polymerase chain reaction (RT-PCR) (C) and enzyme-linked immunosorbent assay (ELISA) (D), respectively. HDs, n = 5; patients, n = 6, with the exception of RT-PCR for TNF expression (n = 5). (E) Immunoblot analysis of ADA2 expression and secretion ADA2+/+ and ADA2−/− U937 macrophages transduced with LV-ADA2 wild-type (ADA2wt) or L188P mutant (ADA2L188P). (F) ADA2 activity was measured in cell-free supernatants from ADA2+/+ and ADA2−/− U937 macrophages transduced with ADA2WT and ADA2L188P. Expression and secretion of IL-6 and TNF were measured in ADA2−/− U937 macrophages expressing ADA2 wild-type or L188P mutant by quantitative RT-PCR (G) and ELISA (H). Data in (F-H) are mean ± standard deviation. *P < .05, **P < .01, ***P < .001. ns, not significant; PT, patient.
Next, we assessed whether macrophage-mediated inflammation is a functional defect caused by loss of ADA2 and whether enzymatic activity acts as an effective brake for macrophage activation, minimizing the risk of unwanted inflammation. To this end, we used a U937 macrophage cell line deficient for ADA2 transduced with an LV encoding wild-type ADA2 (LV-ADA2WT; VCN = 2.10 ± 0.16) or with an L188P mutant ADA2 lacking enzymatic activity (LV-ADA2L188P; VCN = 2.18 ± 0.03) (Figure 7E-F). Upon LPS/IFN-γ exposure, UT ADA2−/− U937 macrophages expressed and secreted IL-6 and TNF at higher levels than did ADA2+/+ macrophages, similarly to patients’ macrophages (Figure 7G-H), confirming that the U937 cell model recapitulates a clinically relevant disease phenotype. The reconstitution of ADA2WT, but not ADA2 L188P, restored physiological secretion of IL-6 and TNF (Figure 7G-H). These data indicate that the amplified inflammatory response of patients’ macrophages is a cell-intrinsic consequence of the loss of ADA2 and that enzymatic activity is essential for the control of macrophage activation.
Discussion
The therapeutic value of allogeneic HSCT for patients with DADA2,27 together with the excellent therapeutic efficacy of gene therapy in patients with severe combined immunodeficiency due to ADA1 deficiency (ADA-SCID),29 suggests that transplantation of autologous gene-corrected HSPCs may also represent a promising strategy for DADA2. Therefore, we designed a preclinical study to evaluate the efficacy of an ADA2-encoding LV in support of its future development for clinical studies. The efficient transduction of myeloid and erythroid progenitors was obtained using LV-ADA2 and optimized gene transfer conditions. ADA2-transduced HDs’ HSPCs expanded as much as did HSPCs transduced with the control vector, and the numbers of CFU-GM and BFU-E colonies derived from ADA2-transduced HSPCs were comparable to the UT condition. HSPC transduction with LV-ADA2 led to a dose-dependent increase in intracellular expression and release of ADA2, which exhibited a proper enzymatic activity. We also used mouse models to examine the effect of ADA2 (over)expression in vivo. Immunocompromised NSGW41 mice were transplanted with ADA2-transduced CD34+ cells isolated from HDs’ BM. LV-ADA2–transduced lineage-negative cells from CD45.1 C57BL/6 mice were also adoptively transferred into congenic C57BL/6 recipients, which are naturally null for ADA2. In both models, efficient engraftment of ADA2-transduced HSPCs was achieved. Human and mouse HSPCs were able to generate a multilineage immune cell repertoire as much as were HSPCs transduced with the GFP control vector. Mice did not show evidence of distress or transduction-related toxicity and exhibited normal spleen and BM tissue architecture. ADA2 overexpression did not change LPS-elicited systemic cytokine response in vivo. These results reveal that ADA2 overexpression is well tolerated in vivo and does not alter the proliferation and maturation potential of myeloid and erythroid progenitors. Additionally, they suggest that tight regulation of ADA2 expression might not be necessary, as also demonstrated in preclinical and clinical studies of ADA-SCID.33,34
An in-depth analysis of the BM compartment of 3 DADA2 patients with hematological manifestations revealed a significant reduction in HSCs and progenitors, which resulted in insufficient production or premature depletion of mature cells of the myeloid, lymphoid, and erythroid lineages. It is unclear whether HSC loss is a direct consequence of ADA2 deficiency in these cells or extrinsic factors (eg, chronic inflammation and autoreactive antibodies) contribute to this phenomenon and eventually cause cytopenia in patients with DADA2. Future studies would be necessary to compare the BM cell composition between children at early disease onset and adults and examine whether anti-TNF therapy prevents or slows HSC loss. Although the absolute number of patients’ HSPCs was low, cell recovery was sufficient to examine the ability of LV-ADA2 to restore ADA2 expression and activity in these cells. Clinically relevant transduction levels (≥75%) and vector copies (1-3 per cell) were achieved in the HSPCs of 2 patients after a single transduction with LV-ADA2. LV-ADA2 reconstituted intracellular ADA2 expression in CD34-derived myeloid cells at normal and supranormal levels after transduction at an MOI of 10 and 50, respectively, which was accompanied by ADA2 secretion in the cell supernatant. Notably, ex vivo transduction and ADA2 overexpression did not alter the ability of patients’ CD34 cells to differentiate in myeloid and erythroid cells. Collectively, these results demonstrate that the LV-ADA2 platform and transduction conditions can efficiently deliver functionally active ADA2 to patients’ HSPCs.
From the perspective of a future clinical application, we examined the ability of our LV-ADA2 to correct the exaggerated release of proinflammatory cytokines from patients’ macrophages. We confirmed that ADA2-deficient macrophages release inflammatory IL-6 and TNF cytokines at high levels in an Italian cohort of patients with a heterogeneous clinical phenotype.1 LV-mediated ADA2 reconstitution in these cells restored physiological secretion IL-6 and TNF. These results were recapitulated in an ADA2-deficient U937 macrophage cell line, indicating that the excessive macrophage inflammatory response is a direct consequence of ADA2 loss rather than a secondary event caused by excessive inflammation occurring in patients. Notably, we demonstrated that the correction of the macrophage inflammatory profile requires ADA2 enzymatic activity.
Several questions need to be answered, including the engraftment level and the conditioning regimen intensity required to achieve disease control, the therapeutic ADA2 range, and the possible selective advantage of transduced cells. HSCT experience shows that full-donor chimerism successfully restored the hematological, immunological, and vascular phenotype in patients with DADA2.26,27 The highly diverse phenotypic variability reported in DADA2 does not always correlate with the residual levels of ADA2 activity.35 Sub optimal ADA2 level seems to be associated with vasculitis, whereas a more extensive loss was observed in patients with hematological disease.36 However, 50% of HD enzymatic activity may be sufficient to revert the clinical manifestations, considering that parents carrying a single allele mutation in the ADA2 gene are typically unaffected. Because a recent study implicated ADA2 in regulating B-cell proliferation and immunoglobulin secretion,20 ADA2 correction in lymphocytes might provide these cells with a growth advantage in patients undergoing HSPC gene therapy. Therefore, a reduced-intensity conditioning regimen may be an option in these patients to overcome the risk of infections and toxicity. Supranormal levels of ADA2 enzymatic activity may eventually compensate for incomplete correction. On the other hand, in patients with a predominant myelomonocytic defect and inflammatory phenotype, a fully myeloablative conditioning regimen may be needed to fully deplete the diseased compartment, because myeloid cells do not usually exhibit a selective advantage, as demonstrated in other myeloid diseases.37 Finally, the ability of corrected HSPCs to engraft in vivo in an inflamed environment following BM failure should be investigated. There may be a need for a bridge treatment with anti-TNF therapy to favor the engraftment of corrected HSPCs. This last measure could also limit posttransplant vascular complications in patients presenting with compromised endothelial integrity caused by DADA2.
Which patients will most likely benefit from an HSPC gene therapy approach for DADA2 once preclinical safety and efficacy studies are completed? Gene therapy may be a promising treatment option for young patients with immunodeficiency and inflammation-associated manifestations or for patients at an initial stage of BM failure syndrome. Indeed, we demonstrated efficient transduction and preserved regenerative potential of HSPCs in 3 adult patients with similar disease characteristics and undergoing anti-TNF therapy at the time of BM harvest. Allogeneic HSCT would remain the most effective treatment for patients, especially young adults and adults, with cytopenias involving single or multiple lineages due to progressive BM failure or reduced regenerative potential of HSPCs. Indeed, these patients may be unable to donate a sufficient amount of autologous HSPCs for gene therapy. However, the use of mobilizing agents, such as granulocyte colony-stimulating factor, alone or in combination with plerixafor, treatment of patients with anti-inflammatory drugs (ie, anti-TNF agents) before HSPC harvest, and improved ex vivo manipulation combining HSPC transduction and expansion may allow for the achievement of clinically relevant doses for HSPC gene therapy for these patients.30,38
In conclusion, we showed the therapeutic value of the LV-ADA2 platform by demonstrating efficient reconstitution of ADA2 expression and activity in patients’ HSPCs and correction of the excessive inflammatory response of patients’ macrophages, which are at the basis of some of the disease manifestations. LV-mediated HSPC gene therapy may represent an effective treatment for patients with DADA2, and further studies are warranted to support its clinical development.
Acknowledgments
The authors thank all of the patients, family members, and staff from all of the clinical units that participated in the study. They thank Claudia Sartirana for assistance with patients’ sample processing, Anna Kajaste-Rudnitski for obtaining the vpl-VPX plasmid, Luca Cesana for preparing the vpl-VPX particles, Rossana Norata for preparing the mouse tissue specimens, Paola Rancoita for some statistical analyses, and the Flow Cytometry Resource, Advanced Cytometry Technical Applications Laboratory for assistance with flow cytometry.
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 841780. This work was supported by the grants E-rare EUROCID project (A.A.), Fondazione Telethon (San Raffaele Telethon Institute for Gene Therapy Core Grant, Project A5) (A.M.), the Jeffrey Modell Foundation, and the Else Kröner Fresenius Prize for Medical Research 2020 (A.A.). The authors thank La Spes organization for supporting fellowships (M.Z. and I.B.).
M.Z. is a PhD candidate at Tor Vergata University (Rome), and this work is submitted in partial fulfillment of the requirement for a PhD.
IRCCS Istituto Giannina Gaslini and IRCCS San Raffaele Hospital are part of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases (Project ID 739543). Selected centers are part of the Italian Onco-Hematology Association and Inborn Error Working Party of The European Society for Blood and Marrow Transplantation.
Authorship
Contribution: A.M., A.A., M.P.C., and L.N. conceived, designed, and supervised the study; M.Z., I.B., F. Barzaghi, S.S., R.J.H., L.B.-R., M.C., and E.P. acquired, analyzed and interpreted data; L.S.S., G.M., P.C., A.L., and S.G. generated and provided study material; F. Sanvito evaluated pathology; F. Barzaghi, F. Schena, S.C., F.C., A.P., M.G., F. Benedetti, M.P.C. provided patient material; P.Y.L. provided enzyme activity data; A.M. wrote the manuscript; and M.Z., I.B., F. Barzaghi, S.S., L.N., M.P.C., and A.A. critically revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alessandra Mortellaro, San Raffaele Telethon Institute for Gene Therapy, IRCCS San Raffaele Scientific Institute, Via Olgettina 60, 20132 Milan, Italy; e-mail: mortellaro.alessandra@hsr.it.

