

Key Points
A patient homozygous for THBD c.793T>A (p.Cys265Ser) developed life-threatening bleeding.
Recombinant human thrombomodulin markedly improved symptoms associated with severe bleeding.

Abstract
We report a 19-year-old Vietnamese woman who experienced several life-threatening bleeding events, including ovarian hemorrhage. Blood analysis revealed a decreased fibrinogen level with markedly elevated fibrinogen/fibrin degradation products and D-dimer levels. Despite hemostatic surgery and administration of several medications, such as nafamostat mesylate, tranexamic acid, and unfractionated heparin, the coagulation abnormalities were not corrected, and the patient experienced repeated hemorrhagic events. We found that administration of recombinant human thrombomodulin (rhTM) remarkably improved the patient’s pathophysiology. Screening and sequencing of the TM gene (THBD) revealed a previously unreported homozygous variation: c.793T>A (p.Cys265Ser). Notably, the Cys265 residue forms 1 of 3 disulfide bonds in the epidermal growth factor (EGF)–like domain 1 of TM. Transient expression experiments using COS-1 cells demonstrated markedly reduced expression of TM-Cys265Ser on the plasma membrane relative to wild-type TM. The TM-Cys265Ser mutant was intracellularly degraded, probably because of EGF-like domain 1 misfolding. The reduced expression of TM on the endothelial cell membrane may be responsible for the disseminated intravascular-coagulation–like symptoms observed in the patient. In summary, we identified a novel TM variant, c.793T>A (p.Cys265Ser). Patients homozygous for this variant may present with severe bleeding events; rhTM should be considered a possible treatment option for these patients.
Introduction
Thrombomodulin (TM) is a single-chain type 1 transmembrane glycoprotein encoded by the TM gene (THBD) and predominantly expressed on the surface of vascular endothelial cells.1 The mature form of human TM has 557 amino-acid residues constituting 10 elements: the N-terminal C-type lectinlike domain, 6 epidermal growth factor (EGF)-like domains, a serine/threonine-rich region, a transmembrane domain, and a short cytoplasmic tail.2–4 The most crucial function of TM is the regulation of coagulation and fibrinolysis. Several other functions, including anti-inflammatory effects through inhibition of complement activity and neutrophil adhesion, have also been described.5
When thrombin binds directly to TM, the thrombin-TM complex accelerates the activation of protein C (PC) more than 1000-fold relative to thrombin alone, and activated PC exerts anticoagulant activity.6,7 The thrombin-TM complex also activates thrombin-activatable fibrinolysis inhibitor (TAFI), inhibiting fibrin degradation catalyzed by plasmin and whose catalytic efficiency is 1250-fold higher than that of thrombin alone.8
Previous animal studies have indicated that a reduction in TM function can trigger thrombotic events. Although complete TM-deficient mice die before birth, the heterozygous null Thbd (TM+/−) allele or mutant Thbd (TMGlu404Pro/−) is not always lethal in mice; however, they produce a hypercoagulable state.9–12 In humans, several genetic polymorphisms in THBD have been reported, with some possibly related to thrombotic diseases, including myocardial infarction, malignant hypertension, and atypical hemolytic uremic syndrome.13–15
Recombinant human soluble TM (rhTM) comprises 498 amino-acid residues derived from TM extracellular domains. rhTM binds to circulating thrombin molecules, inhibiting their procoagulant activity and promoting PC activation.16,17 Besides, it inhibits inflammation and organ injury by degrading the high-mobility group box 1 protein to its proinflammatory form.18 rhTM has been approved in Japan to treat disseminated intravascular coagulation (DIC), associated with sepsis.
In the present study, we found a previously unreported homozygous c.793T>A (p.Cys265Ser) THBD variant in a Vietnamese patient who presented with a hemorrhagic episode of unknown etiology. We describe the cause of coagulation defects in this patient and clarify why rhTM infusion was an effective treatment.
Methods
DNA extraction and PCR amplicon sequencing
Genomic DNA was extracted from whole-blood samples from the proband, her parents, and brother, by using an Illustra blood genomicPrep Mini Spin Kit (Cytiva, Tokyo, Japan). The open reading frame (1728 bp in 1 exon) and flanking regions of THBD were amplified by polymerase chain reaction, with 3 primer pairs (CACTTATAAACTCGAGCCCTGG and AAGTGGAACTCGCAGAGGAAG, CGTCGCTGTCTCCGCTGCTGA and GGCACTGGTACTCGCAGTTG, and CACTGCTACCCTAACTACGACCT and TAAGGTGCTTTGGTAGCAAAGCTG). Polymerase chain reaction products were sequenced in both directions with a Big-Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and an Applied Biosystems 3500xL Genetic Analyzer (Life Technologies, Tokyo, Japan). Written informed consent was obtained from the patient and her family, in agreement with the Declaration of Helsinki.
Construction of expression vectors
The wild-type (WT) THBD coding region was amplified from human genomic DNA and tagged with a C-terminal His tag using Phusion Hot Start Flex 2X Master Mix (New England Biolabs, Ipswich, MA) and cloned into the pCI mammalian expression vector (Promega, Madison, WI). The Cys265-to-Ser (C265S) mutant was generated by site-directed mutagenesis. Sequences were verified using the 3500xL Genetic Analyzer (Applied Biosystems).
Cell culture and transfection
COS-1 cells in 60-mm dishes were transiently transfected with each expression vector by using Lipofectamine 3000 (ThermoFisher Scientific, Waltham, MA). Cells were cultured in Dulbecco’s modified Eagle’s medium (Wako Pure Chemical, Osaka, Japan) supplemented with 10% fetal bovine serum for 16 to 48 hours. Cell membranes were fractionated with the Minute Plasma Membrane Protein Isolation Kit (Invent Biotechnologies, Plymouth, MN). To determine whether the C265S mutant is degraded by endoplasmic reticulum (ER)–associated degradation (ERAD), transfected cells (16 hours) were incubated in 3 µM epoxomicin for 11 hours.
Western blot analysis
Proteins in cell lysates and various fractions were subjected to sodium dodecyl sulfate-polyacrylamide (5%–20% gradient) gel electrophoresis (SDS-PAGE) in reducing conditions and transferred to a polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA). After the reaction was blocked with 5% skim milk, the membrane was incubated with anti-His tag monoclonal antibody-horse radish peroxidase (MBL, Nagoya, Japan) for 2 hours. Chemiluminescence signals were detected and quantified using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA) and a LAS-3000 Imager (Fujifilm, Tokyo, Japan).
PC activation assay
Transfected cells (24 hours) were washed twice with ice-cold assay buffer (20 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid, 150 mM NaCl, 5 mM CaCl2 [pH 7.4]), scraped into 1 mL of assay buffer, pelleted at 3000g for 3 minutes, and gently resuspended in 100 µL of assay buffer. A 10-µL aliquot of the resultant sample was incubated with 0.5 µM human PC (Haematologic Technologies, Essex Junction, VT) and 5 nM human thrombin (Haematologic Technologies) for 30 minutes at 37°C. The reaction was stopped by the addition of 150 µg/mL antithrombin (Haematologic Technologies) and 150 U/mL heparin. The amidolytic effect of activated PC was assayed with 200 µM chromogenic substrate S-2366 (Chromogenix, Milano, Italy), and hydrolysis of S-2366 was quantitated by measuring the absorbance spectrophotometrically at 405 nm. A standard curve was generated using thrombomodulin wild-type (TM-WT), and activity was normalized to total protein levels quantitatively stained in-gel with GelCode Blue Stain Reagent (ThermoFisher Scientific).
TAFI activation assay
Membrane suspensions of transfected cells were prepared as in the PC activation experiments. A 10-µL aliquot of the resultant sample was incubated with 0.375 µM human TAFI and 1.36 nM human thrombin (both from Haematologic Technologies) for 10 min at 37°C. The reaction was stopped by adding 5 µL of 1.6 µM d-phenylalanyl-prolyl-arginyl chloromethyl ketone (Cayman Chemical, Ann Arbor, MI). Then, 10 µL of the reaction mixture was mixed with 5 µL of 30 mM hippuryl-l-arginine (Peptide Institute, Osaka, Japan) and incubated for 45 minutes at 37°C. After the incubation, 100 µL of 0.25 mM phosphate buffer (pH 8.3) and 75 µL of 3% cyanuric chloride in 1.4-dioxane were added and mixed well. The sample was centrifuged at 9100g for 10 minutes. The supernatant (100 µL) was transferred to a 96-well plate, and absorbance at 405 nm was measured. A standard curve was generated using TM-WT, and activity was normalized to total protein levels quantitatively stained in-gel with GelCode Blue Stain Reagent.
Statistical analysis
Data are presented as means ± standard deviation (SD). Student t tests were performed with Prism 8.0 (GraphPad Software, San Diego, CA). A difference of P < .05 was considered to be statistically significant.
Results
Case presentation
A 19-year-old Vietnamese woman developed an acute onset of inferior abdominal pain and was referred to the gynecology department of the hospital of origin. Her parents were not consanguineous, and her mother did not have a spontaneous abortion. She had no family history of bleeding or thrombosis disorders, and neither her family members nor relatives exhibited immunodeficiency symptoms. Further, the patient did not have a history of any chronic inflammatory disease.
Laboratory tests of the patient on the day of referral showed severe anemia, thrombocytopenia, and marked coagulation abnormalities: hemoglobin, 6.3 g/dL; platelet count, 100 × 103/μL ; prothrombin time, 16.5 seconds; activated partial thromboplastin time >150 seconds; fibrinogen, <40 g/dL; fibrinogen/fibrin degradation products (FDPs), 152.9 μg/mL; and D-dimer, 47.6 μg/mL (Table 1 ). A computed tomographic scan of the abdomen and pelvis showed heavy bleeding from the left ovary; subsequently, the patient underwent emergency hemostatic surgery and received a massive blood transfusion.
Laboratory data
| Variable | Reference range | On presentation, referring hospital (day 1) | On presentation, our hospital (day 16) |
|---|---|---|---|
| WBC count (×103/μL) | 4.0-9.0 | 18.2 | 3.9 |
| Hemoglobin (g/dL) | 11.5-15.0 | 6.3 | 10.5 |
| Platelet count (×104/mm3) | 15.0-35.0 | 10.0 | 37.6 |
| PT (s) | 10-13 | 16.5 | 10.2 |
| APTT (s) | 26-40 | >150 | 26.3 |
| Fibrinogen (g/dL) | 160-400 | <40 | 284 |
| Antithrombin III (%) | 80-130 | 68.5 | 68.5 |
| FDP (μg/mL) | 0-5.0 | 152.9 | 115 |
| D-dimer (μg/mL) | 0-1.0 | 47.6 | 45.9 |
| Soluble fibrin monomer complex (FU/mL) | <6.1 | — | >250 |
| Antiplasmin (%) | 80-120 | — | 63 |
| PIC (μg/mL) | <5.0 | — | 21.6 |
| TAT (μg/mL) | 0-3.0 | — | 36.8 |
| PC activity (%) | 70-140 | — | 123 |
| Protein S activity (%) | 65-140 | — | 164 |
| sTM (FU/mL) | 1.8-3.9 | — | >32 |
| Variable | Reference range | On presentation, referring hospital (day 1) | On presentation, our hospital (day 16) |
|---|---|---|---|
| WBC count (×103/μL) | 4.0-9.0 | 18.2 | 3.9 |
| Hemoglobin (g/dL) | 11.5-15.0 | 6.3 | 10.5 |
| Platelet count (×104/mm3) | 15.0-35.0 | 10.0 | 37.6 |
| PT (s) | 10-13 | 16.5 | 10.2 |
| APTT (s) | 26-40 | >150 | 26.3 |
| Fibrinogen (g/dL) | 160-400 | <40 | 284 |
| Antithrombin III (%) | 80-130 | 68.5 | 68.5 |
| FDP (μg/mL) | 0-5.0 | 152.9 | 115 |
| D-dimer (μg/mL) | 0-1.0 | 47.6 | 45.9 |
| Soluble fibrin monomer complex (FU/mL) | <6.1 | — | >250 |
| Antiplasmin (%) | 80-120 | — | 63 |
| PIC (μg/mL) | <5.0 | — | 21.6 |
| TAT (μg/mL) | 0-3.0 | — | 36.8 |
| PC activity (%) | 70-140 | — | 123 |
| Protein S activity (%) | 65-140 | — | 164 |
| sTM (FU/mL) | 1.8-3.9 | — | >32 |
APTT, activated partial thromboplastin time; PIC, plasmin inhibitor complex; PT, prothrombin.
The laboratory findings on the day of admission at the referral hospital (day 1) suggested that the patient had DIC after the hemorrhage. Intravenous nafamostat mesylate and unfractionated heparin were promptly started after surgery to manage DIC; however, these treatments were seemingly ineffective upon laboratory investigation (Figure 1A). On day 6 after admission, the patient developed acute abdominal pain again and was diagnosed with intra-abdominal hemorrhage. Another emergency hemostatic surgery was performed; however, the operation failed to identify the exact source of bleeding, and subsequent venous cauterization also failed to stop the bleeding completely. The bleeding was eventually stopped using massive fresh frozen plasma (FFP) and concentrated platelet transfusion.

Blood investigation trends. (A) Changes in levels of key blood factors after massive FFP transfusion and hemostatic surgery on day 1 (D1). The patient had a second intra-abdominal hemorrhage on day 6 (D6), requiring another emergency hemostatic surgery. Eighteen units of FFP were also administered after the hemorrhage. (B) Changes in fibrinogen, FDP, and D-dimer levels after rhTM infusions (380 U/kg) administered IV once daily from days 7 through 9 and days 11 through 14. After rhTM infusions were stopped, nafamostat mesylate and tranexamic acid (TA) were administered beginning on day 16. Large FFP transfusions occurred daily from days 17 through 20. (C) The effect of continued nafamostat mesylate and TA treatment followed by reinfusion with rhTM on the levels of coagulation factors over time. On day 36, the daily dosing interval of rhTM was extended to every 2 to 3 days. (D) Changes in levels of key blood coagulation factors followed by infusion with rhTM from days 29 through 34. UFH, unfractionated heparin.
Blood investigation trends. (A) Changes in levels of key blood factors after massive FFP transfusion and hemostatic surgery on day 1 (D1). The patient had a second intra-abdominal hemorrhage on day 6 (D6), requiring another emergency hemostatic surgery. Eighteen units of FFP were also administered after the hemorrhage. (B) Changes in fibrinogen, FDP, and D-dimer levels after rhTM infusions (380 U/kg) administered IV once daily from days 7 through 9 and days 11 through 14. After rhTM infusions were stopped, nafamostat mesylate and tranexamic acid (TA) were administered beginning on day 16. Large FFP transfusions occurred daily from days 17 through 20. (C) The effect of continued nafamostat mesylate and TA treatment followed by reinfusion with rhTM on the levels of coagulation factors over time. On day 36, the daily dosing interval of rhTM was extended to every 2 to 3 days. (D) Changes in levels of key blood coagulation factors followed by infusion with rhTM from days 29 through 34. UFH, unfractionated heparin.
After the second surgery, rhTM, was administered IV in doses of 380 U/kg once daily for 1 week, leading to a marked improvement of the hemostatic profile: specifically, increased fibrinogen levels and decreased FDP and D-dimer levels (Figure 1B). However, after rhTM withdrawal on day 14, the laboratory coagulation profile deteriorated. Therefore, the patient was diagnosed with a hematological anomaly of undefined origin and was transferred to Tokyo Saiseikai Central Hospital on day 16 for further evaluation.
On admission to our hospital, laboratory analysis revealed low fibrinogen levels with markedly elevated FDP and D-dimer levels (Figure 1B). More in-depth coagulation examination showed a high plasmin-α2-plasmin-inhibitor complex (21.6 μg/mL) with decreased antiplasmin activity (63%), suggesting severe fibrinolysis (Table 1 ). The patient reported that, during her childhood, bleeding was difficult to stop after injury.
We administered tranexamic acid and nafamostat mesylate from day 16. However, neither drug was effective; thus, a large FFP transfusion was administered. These treatments were discontinued on day 28, owing to the appearance of deep vein thrombosis in the legs. Although fibrinogen concentrate had been tested several times, it did not improve the abnormal coagulation. Owing to the apparent benefit of rhTM treatment at the referring hospital, we restarted daily rhTM infusion on day 30, which produced a marked improvement in coagulation status (Figure 1C-D). These results suggested that rhTM was the only irreplaceable therapeutic agent for this patient; thus, we continued to administer rhTM intermittently to maintain her fibrinogen level.
On day 42, we started to extend the daily interval of rhTM treatments to 2 to 3 per day with daily laboratory monitoring; however, on day 76, the patient had sudden abdominal pain and went into shock. She was diagnosed with massive bleeding from the right ovary; thus, an emergency ovariectomy for hemostasis was successfully performed. After this bleeding event, the patient received rhTM infusions every 2 to 3 days to maintain her blood fibrinogen level above 200 mg/dL.
A novel, rare THBD variation and a common polymorphism
Two missense variants, c.793T>A (p.Cys265Ser) and c.1418C>T (p.Ala473Val), were identified in THBD of the proband (Figure 2). To the best of our knowledge, the c.793T>A variant has not been reported. The c.1418C>T variant is a common polymorphism with an allele frequency of ∼15% and 29%, worldwide and in East Asian populations, respectively, according to the public database gnomAD v3.19
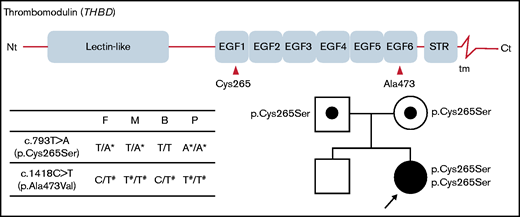
Structure of the thrombomodulin gene (THBD) and identification of 2 missense variants in the patient and her parents. The c.793T>A substitution causes the p.Cys265Ser missense substitution in EGF-like domain 1, and c.1418C>T causes p.Ala473Val in EGF-like domain 6. The patient’s parents were both heterozygous for the c.793T>A variant, whereas her brother did not carry it. Nt, N terminus; STR, Ser/Thr-rich domain; tm, transmembrane domain; Ct, C terminus; F, father; M, mother; B, brother; P, proband. *Novel variation. #Common polymorphism.
Structure of the thrombomodulin gene (THBD) and identification of 2 missense variants in the patient and her parents. The c.793T>A substitution causes the p.Cys265Ser missense substitution in EGF-like domain 1, and c.1418C>T causes p.Ala473Val in EGF-like domain 6. The patient’s parents were both heterozygous for the c.793T>A variant, whereas her brother did not carry it. Nt, N terminus; STR, Ser/Thr-rich domain; tm, transmembrane domain; Ct, C terminus; F, father; M, mother; B, brother; P, proband. *Novel variation. #Common polymorphism.
The patient’s parents were both heterozygous for the c.793T>A variant, suggesting that the variant alleles were inherited. Her brother did not carry the c.793T>A variant. As the symptomatic proband was the only homozygote for c.793T>A in the family, this is a candidate for the cause of the recessive coagulation disorder of the proband.
C265S reduced cell surface expression
To assess the effect of c.793T>A (p.Cys265Ser) substitution on TM expression, we expressed recombinant TM-WT and -C265S proteins in COS-1 cells and performed western blot analysis (Figure 3A). In total cell lysates, TM-WT proteins were observed as an upper main band (∼90 kDa) and lower band (∼80 kDa), presumably representing the mature (glycosylated) and nonglycosylated forms, respectively (Figure 3A lane 2). The C265S mutant exhibited a reduced level of the top band and an increased level of the bottom band (Figure 3A lane 3), suggesting that a substantial fraction of the mutant TM protein could not be converted to or remained in the mature form. Cell fractionation revealed that plasma membrane expression of TM in the C265S mutant was significantly reduced (24.2% ± 14.3%; P = .012) relative to that in the WT (Figure 3A lanes 11, 12; Figure 3B).
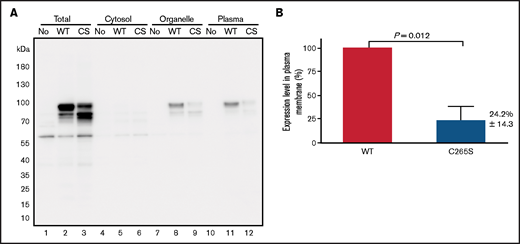
Expression of the TM C265S mutant is reduced on the cell surface. (A) Representative TM western blot data from total cell lysates (Total), cytosol, organelle membranes (Organelle), and plasma membrane (Plasma) fractions prepared from nontransfected cells (No) and cells transfected with TM-WT or -C265S mutant (CS) expression vectors. His-tagged TM protein was detected after SDS-PAGE and western blot analysis, with an anti-His tag antibody. The top main band at ∼90 kDa and the bottom one at ∼80 kDa presumably represent the mature glycosylated and unglycosylated forms of TM, respectively. (B) Quantification of TM in plasma membrane fractions of WT or C265S cells (n = 3 experiments; mean ± SD).
Expression of the TM C265S mutant is reduced on the cell surface. (A) Representative TM western blot data from total cell lysates (Total), cytosol, organelle membranes (Organelle), and plasma membrane (Plasma) fractions prepared from nontransfected cells (No) and cells transfected with TM-WT or -C265S mutant (CS) expression vectors. His-tagged TM protein was detected after SDS-PAGE and western blot analysis, with an anti-His tag antibody. The top main band at ∼90 kDa and the bottom one at ∼80 kDa presumably represent the mature glycosylated and unglycosylated forms of TM, respectively. (B) Quantification of TM in plasma membrane fractions of WT or C265S cells (n = 3 experiments; mean ± SD).
Plasma membrane proteins, including TM, are synthesized and folded in the ER. The cellular ERAD system degrades unfolded or misfolded proteins accumulated in the ER. When cells expressing the TM C265S mutant were treated with the selective proteasome inhibitor epoxomicin, the lower molecular weight TM band further accumulated (Figure 4 lane 2), especially in the organelle fraction (lane 6). Thus, the C265S variant TM was likely to be degraded by ERAD, probably because of the misfolding of the EGF-like domain 1 containing the substituted residue.
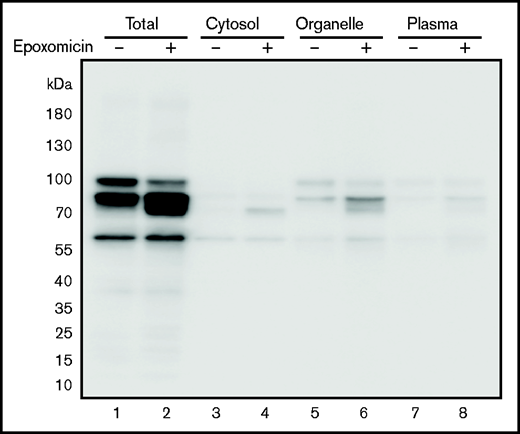
TM levels after treatment of cells with epoxomicin. Cells expressing the TM-C265S mutant protein were cultured in the absence or presence of the proteasome inhibitor epoxomicin for 11 hours. TM protein was subsequently detected after separation by SDS-PAGE and western blot analysis with an anti-His tag antibody. Total, cell lysates; Organelle, organelle membranes; Plasma, plasma membranes.
TM levels after treatment of cells with epoxomicin. Cells expressing the TM-C265S mutant protein were cultured in the absence or presence of the proteasome inhibitor epoxomicin for 11 hours. TM protein was subsequently detected after separation by SDS-PAGE and western blot analysis with an anti-His tag antibody. Total, cell lysates; Organelle, organelle membranes; Plasma, plasma membranes.
To assess the functional impact of the C265S mutation, the PC and TAFI activation activities of TM-WT and -C265S proteins were assayed by using transfected cell membranes. The PC activation activity of C265S on the cell surface was significantly reduced (34.9% ± 7.7%; P = .0013) relative to that of the WT (Figure 5A). The TAFI activation activity of C265S on the cell surface was also significantly reduced (27.6% ± 10.1%; P = .0010; Figure 5B). These data were consistent with the results showing that the expression of C265S on the plasma membrane was reduced (Figure 3). The data also suggested that the C265S mutant protein on the cell surface retains its native specific activity as a cofactor in the thrombin-mediated PC and TAFI activation. Under the experimental condition, nontransfected cells were not detected in PC and TAFI activation assays (data not shown).
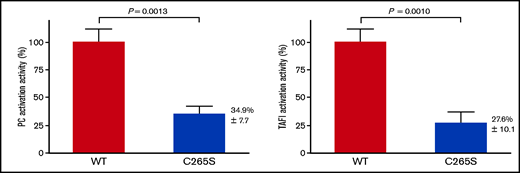
Activation of PC and thrombin-activatable fibrinolysis inhibitor (TAFI) by thrombin and cells expressing TM-WT or the TM-C265S mutant. (A) Cells expressing WT or C265S mutant TM were incubated with PC and thrombin at 37°C for 30 minutes before stopping the reaction by the addition of antithrombin and heparin. The amidolytic activity of activated PC was assayed using S-2366 (n = 3 experiments; mean ± SD). Nontransfected cells showed no detectable activity. (B) Cells expressing WT or C265S mutant TM were incubated with TAFI and thrombin at 37°C for 10 minutes before stopping the reaction by the addition of d-phenylalanyl-prolyl-arginyl chloromethyl ketone. The carboxypeptidase activity of activated TAFI was assayed using hippuryl-l-arginine (n = 3 experiments; mean ± SD). Nontransfected cells showed no detectable activity.
Activation of PC and thrombin-activatable fibrinolysis inhibitor (TAFI) by thrombin and cells expressing TM-WT or the TM-C265S mutant. (A) Cells expressing WT or C265S mutant TM were incubated with PC and thrombin at 37°C for 30 minutes before stopping the reaction by the addition of antithrombin and heparin. The amidolytic activity of activated PC was assayed using S-2366 (n = 3 experiments; mean ± SD). Nontransfected cells showed no detectable activity. (B) Cells expressing WT or C265S mutant TM were incubated with TAFI and thrombin at 37°C for 10 minutes before stopping the reaction by the addition of d-phenylalanyl-prolyl-arginyl chloromethyl ketone. The carboxypeptidase activity of activated TAFI was assayed using hippuryl-l-arginine (n = 3 experiments; mean ± SD). Nontransfected cells showed no detectable activity.
Discussion
The patient claimed that bleeding had been difficult to stop when injured on several occasions throughout her childhood. These experiences suggested that congenital or genetic problems caused her frequent bleeding episodes. A mutation in fibrinogen genes was suspected initially but seemed unlikely, because her laboratory data showed no response to transfusion of FFP or fibrinogen concentrate. There were also no data suggesting congenital coagulation diseases, such as coagulation factor deficiency and von Willebrand disease. Because rhTM appeared to be effective in treating her disease, we hypothesized the existence of genetic anomalies in her THBD. Indeed, we discovered an unreported THBD variation, c.793T>A (p.Cys265Ser). We also found that both of her parents were heterozygous for this variant, but that it was absent in her brother. From these genetic data and the marked improvement in the coagulation abnormality with rhTM administration, we concluded that the several massive bleeding events that she had experienced were linked to the C265S variant homozygosity. Of note, based on the absence of hemorrhagic events in the parents, this allele appeared to be recessive.
Although we failed to measure her soluble TM (sTM) levels at baseline, we have data from day 40, 4 days after the latest rhTM infusion. Considering the pharmacokinetics (Cmax, 807 ng/mL; t1/2, 16.2 hours) of rhTM in patients without renal dysfunction, her sTM level at day 40 (>32 FU /mL) was considered to include not only infused rhTM (estimated to be ∼0.08 FU/mL) but also her endogenous TM. Although the exact trigger that caused the ectodomain shedding of endogenous TM is unknown, it could be due to a certain inflammatory state latent in the patient.20
Results from previous studies with Thbd-null mice indicated that homozygosity for a Thbd-null allele was fatal9 ; however, this was not the case for our patient with the homozygous THBD variant. We observed partial expression of the TM-C265S mutant on the cell surface in vitro. These results indicated that this variant remained functionally normal to some extent.
Among the several structural consequences of TM, its anticoagulation and antifibrinolysis activities are primarily associated with EGF-like domains. For instance, the thrombin anion-binding exosite 1 binds to EGF5-6 of TM. EGF4-6 is essential and sufficient for PC cleavage and activation,1,21,22 whereas TM activity for TAFI activation resides in EGF3-6.2,23 The biological and clinical significance of the EGF1 domain, including Cys265, has yet to be elucidated. Given that the replaced residue is not associated with EGF3-6, the C265S mutant is likely to activate PC and TAFI normally once expressed on the cell surface. In our investigation, the cell surface expression of the TM-C265S mutant decreased because of its degradation by ERAD, concomitantly reducing its ability to activate PC and TAFI. The decreased activation of PC may have caused this patient’s deep vein thrombosis, and the decreased activation of TAFI may have caused a hyperfibrinolytic state, leading to some of her hemorrhagic events.
Mice carrying a homozygous Glu404Pro mutation, located in the loop between the EGF4 and EGF5 domains, can be born normally, although there is a 1000-fold reduction in TM activation of PC compared with that in WT mice.24 On the other hand, 40% of TMLoxP/lacZCretg-carrying mice die in the embryonic phase because of placental defects.25,26 According to Isermann et al,27 placental defects in TM-deficient mice were caused by cell death and growth inhibition of placental trophoblast cells. They stated that not only fibrin, but also subsequent FDP may lead to the death of giant trophoblast cells. Reduced TAFI activation ability in TM-deficient mice may cause more fibrin degradation, resulting in placental defects.
In contrast to these mouse models, the clinical symptoms of the patient in this study were characterized by recurrent hemorrhage, indicating that TM-C265S mainly causes hyperfibrinolysis rather than hypercoagulation. The C265S mutation may disrupt the timely and delicate balance between coagulation and fibrinolysis.
Ovulation may have triggered her bleeding because the bleeding occurred when her basal body temperature rose. However, it is hard to know why the bleeding became severe on this occasion. The corpus luteum becomes cystic and filled with blood after ovulation. Rupture of the corpus luteum is rare in healthy women but can sometimes cause life-threatening intraperitoneal hemorrhage in women taking anticoagulants or having bleeding disorders.28 Although infertility treatment, sexual intercourse, ovarian cysts, or polyps can trigger severe ovulation bleeding, this patient had none of them. In the ovary, TM reportedly functions in the optimal luteinization of preovulatory follicles by mediating thrombin and protease-activated receptors (PAR1 and PAR4).29 The TM C265S mutation in this patient may affect not only the balance between coagulation and fibrinolysis but also other biological functions, such as intracellular signaling pathways, through PAR1/PAR4 receptors.
A case with different homozygous THBD variants was recently reported by Okada et al.30 In their report, a 6-month-old male patient with a homozygous TM-Gly412Asp variant developed recurrent subcutaneous bleeding related to coagulation-fibrinolysis system abnormalities, which were markedly improved by rhTM infusion. Unlike our patient, the TM-Gly412Asp missense variation was in the EGF5 domain of TM, producing a lack of thrombin binding and failing to activate PC and TAFI in vitro. These findings may explain why this patient’s bleeding symptoms occurred earlier during childhood and appeared to be more severe than those of our patient.
Two other THBD variants were reported to be associated with bleeding: c.1611C>A (p.Cys537*)31 and c.1487delC (p.Pro496Argfs*10).32 Heterozygous patients with these variants showed continuously elevated sTM levels in the plasma related to incomplete C-terminal structures of TM, resulting in reduced thrombin generation and bleeding events. In contrast, patients with the c.793T>A (p.Cys265Ser) variant showed symptoms only when homozygous, not when heterozygous. It is unlikely that the Cys265 mutation in EGF1 enhances the shedding of TM-like mutations of Cys537 in the transmembrane domain and of Pro496 in the serine/threonine-rich region. The elevation of sTM in the patient with p.Cys265Ser may not be the cause of the bleeding symptoms, but rather the result of endothelial damage related to imbalance of the coagulation-fibrinolysis system associated with reduced expression of endothelial TM. In fact, rhTM infusion was very effective for this patient. A limitation of this study is that we did not have plasma samples of the patient before the rhTM infusion, so we could not confirm that sTM was not constitutively elevated in the absence of symptoms.
In summary, we have identified a previously unreported THBD variant, c.793T>A (p.Cys265Ser). A patient homozygous for c.793T>A (p.Cys265Ser) presented with severe bleeding that was controlled with rhTM infusion. The decreased cell surface expression of the TM-C265S variant may tilt the balance between coagulation and fibrinolysis. A TM disorder should be investigated when bleeding of uncertain etiology occurs.
Acknowledgments
The authors thank Nguyen Thi Mai of the National Institute of Hematology and Blood Transfusion, Hanoi, for clinical management of the patient in Vietnam.
This work was supported in part by JSPS.
Asahi Kasei Pharma provided the Recomodulin (rhTM) for compassionate use.
Authorship
Contribution: M.O. was in charge of the clinical management of the patient, designed the study, and wrote the manuscript; K.M. and K.K. designed the study, performed the research, and wrote the manuscript; R.D., K.Y., H.K., and M.H. were in charge of the clinical management of the patient and reviewed the manuscript; S.M., N.O., M.M., and Y.I. helped perform experiments, analyze the data, and reviewed the manuscript; and Y.T. and T.K. were in charge of the clinical management of the patient, designed the study, and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Makoto Osada, Department of Hematology, Tokyo Saiseikai Central Hospital, 1-4-17 Mita, Minato-ku, Tokyo 108-0073, Japan; e-mail: makoosd.0221@gmail.com.

