

Key Points
Effector polyfunctional index score of CD4 cells was more associated with responses to combined azacitidine/nivolumab than CD8 cells.
Single-cell biomarker assays can be used in predicting responses to immune-based therapies.
Abstract
Acute myeloid leukemia (AML) remains a difficult disease to treat disease. In a phase 2 clinical trial in patients with relapsed/refractory AML, combining the hypomethylating agent, azacitidine, with the PD-1 checkpoint inhibitor, nivolumab, demonstrated encouraging response rates (33%), median event-free, and overall survival, compared with a historical cohort of contemporary patients treated with azacitidine-based therapies, with an acceptable safety profile. Biomarkers of response are yet to be determined. In this study, we leveraged a multiplexed immune assay to assess the functional states of CD4+ and CD8+ cells at a single-cell level in pretherapy bone marrows in 16 patients with relapsed/refractory AML treated with azacitidine/nivolumab. Effector CD4+ but not CD8+ cells had distinct polyfunctional groups and were associated with responses and better outcomes. Further evaluation of the polyfunctional strength index composition across cell types revealed that interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) were the major drivers of enhanced polyfunctionality index of pretherapy CD4+ subset, whereas Granzyme B, IFN-γ, MIP-1b, and TNF-α drove the nonsignificantly enhanced pretreatment Polyfunctional Strength Index of CD8+ subset in the responders. Single-cell polyfunctional assays were predictive of response in AML and may have a potential role as a biomarker in the wider sphere of immunotherapy.
Introduction
Acute myeloid leukemia (AML) remains a difficult disease to treat with a limited proportion of patients having long-term cures, despite several recently US Food and Drug Administration–approved therapies.1 T cells can be used to eradicate leukemia as evident by grafted allogeneic T cells vs leukemia effected seen in postallogeneic stem cell transplantation.2 Harnessing the host T-cell activity against tumors via T-cell checkpoint inhibition has demonstrated significant success in many solid cancers and some lymphomas.3 Checkpoint blockade via PD-1, PD-L1, CTLA-4, and TIM-3 inhibition is being evaluated in several ongoing clinical trials in AML and myelodysplastic syndrome.4 In a phase 2 clinical trial in patients with relapsed/refractory (R/R) AML, combining the hypomethylating agent (HMA), azacitidine, with the PD-1 checkpoint inhibitor, nivolumab, demonstrated encouraging overall response rates of 33% (58% and 22% in HMA-naïve and HMA-pretreated patients), median event-free, and overall survival (OS), compared with a historical cohort of contemporary patients treated with azacitidine-based therapies, with an acceptable safety profile.5 Of note, patients who achieved complete response (CR)/incomplete blood count recovery (CRi)/partial response/hematologic improvement/stable disease had significantly improved OS compared with nonresponders (16.2 vs 4.1 months).5 However, unlike in solid tumors, long-term responses were rarely seen with AML. Patients with increased pretherapy bone marrow CD3+ or CD3+ CD8+ T cells were significantly more likely to achieve a response suggesting that biomarker-based strategies may further improve outcomes with checkpoint blockade in AML. However, bone marrow T-cell infiltration does not take into account the functional status of the T cells. In this study, we leveraged a multiplexed immune assay to assess the functional states of CD4+ and CD8+ cells at a single-cell level in pretherapy bone marrows in patients with R/R AML treated with azacitidine/nivolumab in the aforementioned clinical trial.
Methods
Patient population
All patients were treated on protocol with azacitidine/nivolumab, as previously described (NCT02397720).5 This study was approved by MD Anderson institutional review board and conducted in accordance with the Declaration of Helsinki. Briefly, 11 nonresponders and 5 responders to treatment based on ELN2017 response criteria6 had available samples and were analyzed. Bone marrow samples were collected before and at end of second treatment cycle. Patient clinical and treatment characteristics are included in supplemental Table 1.
Single-cell 32-plex functional proteomic profiling
Detailed methods can be found in supplemental Material under “Single-cell 32-plex functional proteomic profiling.” Briefly, polyfunctional T cells co-secreting 2+ cytokines were assessed with the 32-plex proteomics (supplemental Table 2). The Polyfunctional Strength Index (PSI) of single T cells was computed as the percentage of polyfunctional cells, multiplied by the sum of the mean fluorescence intensity of the proteins secreted by those cells.7-12
Results and Discussion
A total of 16 patients with R/R AML (75% male), 81% with >1 salvage, median age 65 years (range, 47-90), who received a median of 3 (range, 2-17) cycles and a median of 6 (range, 3-33) doses of nivolumab had available samples for this analysis. Five of 16 (31.2%) patients achieved a CR/CRi on azacitidine/nivolumab, with a median time to CR/CRi of 4.07 (range, 0.9-12.6) months. The median OS of the 16 patients was 6 (range, 2.4-21.5) months, (responders vs nonresponders, P < .0001). The median duration of response was 5.2 (range, 0.5-14.7) months.
To identify predictors of response, we conducted single-cell polyfunctional assessment of CD4+ and CD8+ T cells in pretreatment bone marrows. We measured the degree of polyfunctionality and PSI, representing an aggregate of all single-cell multidimensional data into a single index, to determine any association with responses. The pretreatment bone marrow CD4+ but not CD8+ T cells had significantly higher frequency of polyfunctional cells (P = .04 and .15, respectively) and significantly higher PSI (P = .01 and .18, respectively) in patients who achieved CR/CRi compared with nonresponders (Figure 1A-B). Also, pretreatment bone marrow PSI of CD4+ (P < .001) but not CD8+ (P = .14) subsets demonstrated a significant positive association with OS across all patients, likely partly driven by response enrichment, suggesting that the degree of polyfunctional state of CD4+ cells may potentially also predict outcomes in patients with AML treated with azacitidine/nivolumab (Figure 1C). Further evaluation of the PSI composition across cell types revealed that interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) were the major drivers of enhanced PSI of pretherapy CD4+ subset, whereas Granzyme B, IFN-γ, MIP-1b, and TNF-α drove the nonsignificantly enhanced pretreatment PSI of CD8+ subset in the responders (Figure 1D). Although we do not have further subtyping of CD4+ cells, the increased IFN-γ and TNF-α pattern is generally distinctive for the CD4+ TH1 functional state.13
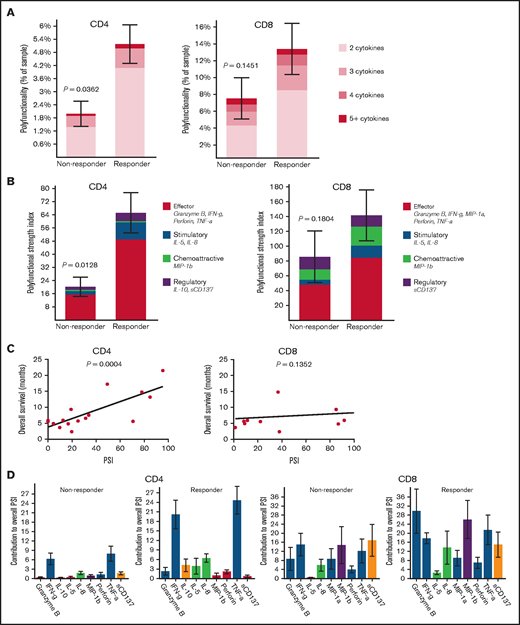
Pretreatment polyfunctional bone marrow CD4+ T cells demonstrate the significant positive correlations with AML patient response to the anti–PD-1–based therapy and overall survival. Polyfunctionality, defined as single-cells co-secreting at least 2 or more proteins, reveals the varying degrees of polyfunctional single cells from the samples, with the darker the orange, the higher the number of unique cytokine combinations secreted per single-cell. PSI aggregates all single cell multidimensional data into a single index, defined as the percentage of polyfunctional cells, multiplied by the sum of the mean fluorescence intensity of the proteins secreted by those cells.7–10,19,21,22 The displayed index is color-coded to show the contribution from different categories of cytokines (eg, effector, stimulatory, chemoattractive, regulatory cytokines). (A) Polyfunctional CD4+ subset can significantly distinguish responding patients to the therapy from nonresponding patients compared with CD8+ subset. (B) PSI of CD4+ subset can significantly segregate responding patients to the therapy from nonresponding patients compared with CD8+ subset, with greater increases of antitumor-associated protein secretions in both CD4+ and CD8+ subsets from responders relative to nonresponders. (C) PSI of CD4+ subset shows a significant positive association with OS (overall survival in months) compared with CD8+ PSI, suggesting the impact of polyfunctional bone marrow CD4+ T-cell subsets on predicting AML patient responses to anti–PD-1 therapy. (D) The PSI composition further uncovers IFN-γ and TNF-α are the major drivers for enhanced PSI of CD4+ subset dissecting the response differences to the therapy between responding and nonresponding patients, whereas Granzyme B, IFN-γ, MIP-1b, and TNF-α seem to mainly drive enhanced PSI of CD8+ subset in the responders. PSI profiles were broken down per cytokine, between response groups, to reveal the specific proteins driving the PSI. The statistical P values were computed using Mann-Whitney U test or Pearson's correlations.
Pretreatment polyfunctional bone marrow CD4+ T cells demonstrate the significant positive correlations with AML patient response to the anti–PD-1–based therapy and overall survival. Polyfunctionality, defined as single-cells co-secreting at least 2 or more proteins, reveals the varying degrees of polyfunctional single cells from the samples, with the darker the orange, the higher the number of unique cytokine combinations secreted per single-cell. PSI aggregates all single cell multidimensional data into a single index, defined as the percentage of polyfunctional cells, multiplied by the sum of the mean fluorescence intensity of the proteins secreted by those cells.7–10,19,21,22 The displayed index is color-coded to show the contribution from different categories of cytokines (eg, effector, stimulatory, chemoattractive, regulatory cytokines). (A) Polyfunctional CD4+ subset can significantly distinguish responding patients to the therapy from nonresponding patients compared with CD8+ subset. (B) PSI of CD4+ subset can significantly segregate responding patients to the therapy from nonresponding patients compared with CD8+ subset, with greater increases of antitumor-associated protein secretions in both CD4+ and CD8+ subsets from responders relative to nonresponders. (C) PSI of CD4+ subset shows a significant positive association with OS (overall survival in months) compared with CD8+ PSI, suggesting the impact of polyfunctional bone marrow CD4+ T-cell subsets on predicting AML patient responses to anti–PD-1 therapy. (D) The PSI composition further uncovers IFN-γ and TNF-α are the major drivers for enhanced PSI of CD4+ subset dissecting the response differences to the therapy between responding and nonresponding patients, whereas Granzyme B, IFN-γ, MIP-1b, and TNF-α seem to mainly drive enhanced PSI of CD8+ subset in the responders. PSI profiles were broken down per cytokine, between response groups, to reveal the specific proteins driving the PSI. The statistical P values were computed using Mann-Whitney U test or Pearson's correlations.
Although checkpoint blockade-based responses in solid tumors are generally thought to be driven by CD8+ cells, recent studies suggest that CD4+ cells are critical in initiating and sustaining an immunotherapy response.14,15 Further, CD4+ cells can independently recognize mutant neoepitopes,16 tumor infiltrating TH1-like CD4+ T cells can acquire antitumor cytotoxic activity,17 and CD4+ rather than CD8+ T cells are critical for inducing responses to neoantigen vaccination in melanoma.18 Collectively, these studies support a significant role for CD4+ in induction of antitumor immunotherapy-based responses, which is consistent with our findings in AML.
We next applied 3-dimensional t-distributed stochastic neighbor embedding19-21 to visualize high-dimensional data in low-dimensional space to stratify functional cell clustering with distinct cytokine secretion patterns between patient groups (Figure 2A). Our findings revealed that CD4+ and CD8+ cells from responders and nonresponders had distinct clustering patterns (Figure 2A, left). However, responders demonstrated a more prominent increase in polyfunctional cell subsets with antitumor-associated proteins in CD4+ and CD8+ subsets (purple circles), compared with nonresponders (Figure 2A, middle and left). When clustered by degree of polyfunctionality, highly polyfunctional groups clustered together in both CD4+ and CD8+ T cells (Figure 2A, middle). Of note, the most dominant functional group across CD4+ and CD8+ cells was the effector protein pattern, which was more notable in the pretreatment bone marrows of responders compared with nonresponders (Figure 2A, left and right) and also corresponded to the highly polyfunctional group. These findings suggest that an effector functional phenotype is the predominant CD4+ and CD8+ T-cell phenotype in pretreatment bone marrows of patients with AML who responded to azacitidine/nivolumab. In addition, we used a functional heatmap to compare the frequency of various expressed monofunctional and polyfunctional groups.7,19,21,22 Compared with nonresponders, the pretherapy CD4+ T cells of responders had more distinct and unique polyfunctional groups (green rectangles) such as coexpression of TNF-α/IFN-γ/interleukin-8, TNF-α/IFN-γ/Granzyme B or TNF-α/IFN-γ/Perforin/interleukin-8 proteins (Figure 2B). However, CD8+ T cells did not show such distinct patterns between responders and nonresponders. These observations are consistent with the pretherapy PSI patterns noted in CD4+ T cells but not CD8+ T cells of responders, further highlighting the association of bone marrow polyfunctional CD4+ subset with response to azacitidine/nivolumab therapy.
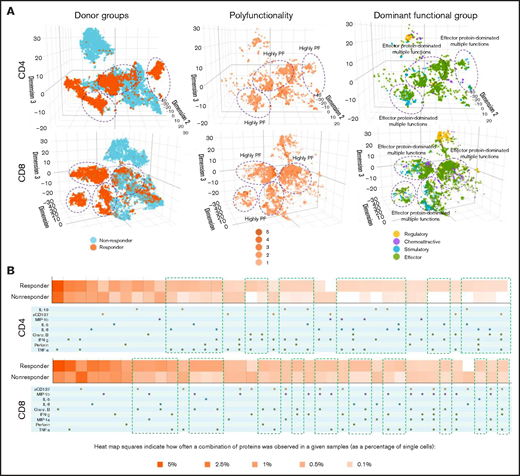
Three-dimensional t-distributed stochastic neighbor embedding (t-SNE) and heatmap visualizations reveal distinct cell clusters with cytokine signatures in pretreatment bone marrow CD4+ and CD8+ cell subsets between nonresponding and responding patients to the anti–PD-1–based therapy. (A) Three-dimensional t-SNE functional graphs plot single cells by differentiating them based on their greatest cytokine-based functional differences. The responders show more prominent increases of polyfunctional cell subsets with antitumor-associated protein secretions in CD4+ and CD8+ subsets than nonresponders (highlighted in purple circles). Donor group cell mapping stratifies data points from samples by responders (orange) and nonresponders (blue). Polyfunctionality cell mapping visualizes the data points based on the degree of polyfunctionality from the sample, with the darker the orange, the higher number of unique cytokines secreted per single cell. Dominant functional group mapping displays a color-coded visualization of data points based on the dominant cytokine profile being secreted, revealing biological drivers. (B) Functional heatmap compares the frequency at which various monofunctional and polyfunctional groups are secreted by the samples. The heatmap reveals the greater upregulation of polyfunctional subpopulations with unique cytokine signatures in both CD4+ and CD8+ subsets from responding patients compared with nonresponders (highlighted in green rectangles).
Three-dimensional t-distributed stochastic neighbor embedding (t-SNE) and heatmap visualizations reveal distinct cell clusters with cytokine signatures in pretreatment bone marrow CD4+ and CD8+ cell subsets between nonresponding and responding patients to the anti–PD-1–based therapy. (A) Three-dimensional t-SNE functional graphs plot single cells by differentiating them based on their greatest cytokine-based functional differences. The responders show more prominent increases of polyfunctional cell subsets with antitumor-associated protein secretions in CD4+ and CD8+ subsets than nonresponders (highlighted in purple circles). Donor group cell mapping stratifies data points from samples by responders (orange) and nonresponders (blue). Polyfunctionality cell mapping visualizes the data points based on the degree of polyfunctionality from the sample, with the darker the orange, the higher number of unique cytokines secreted per single cell. Dominant functional group mapping displays a color-coded visualization of data points based on the dominant cytokine profile being secreted, revealing biological drivers. (B) Functional heatmap compares the frequency at which various monofunctional and polyfunctional groups are secreted by the samples. The heatmap reveals the greater upregulation of polyfunctional subpopulations with unique cytokine signatures in both CD4+ and CD8+ subsets from responding patients compared with nonresponders (highlighted in green rectangles).
The single-cell multiplexed functional proteomics precision profiling demonstrated that the pretreatment PSI of CD4+ cells was significantly associated with response and OS in relapsed patients treated with azacitidine/nivolumab and should be evaluated prospectively to ascertain whether this could be an effective biomarker to select AML patients for PD-1–based therapies. Pretreatment PSI should be evaluated in association with treatment outcomes with CTLA4, PD-L1, TIM3, and other T-cell based strategies in clinical trials in AML and may have a potential role as a biomarker in the wider sphere of immunotherapy in AML. Such analyses are warranted and encouraged.
Acknowledgments
This work was supported in part by the MD Anderson Cancer Centre Support Grant CA016672, the MD Anderson Cancer Center Leukemia SPORE CA100632, the Charif Souki Cancer Research Fund, the Dick Clark Immunotherapy Fund, and generous philanthropic contributions to the MD Anderson Moon Shots Program.
Authorship
Contribution: H.A.A., S.M., M.C., J.Z., M. Andreeff, M.K., and N.D. designed the study, analyzed the data, and wrote the paper; H.A.A., J.M., G.C.I., M. Alfayez, S.M., M.C., and J.Z. collected and analyzed the data; G.C.I. and M. Andreeff performed the molecular and cytogenetic analysis; N.D., M. Andreeff, E.J., S.M.K., M.K., and G.G.-M. enrolled patients; and all authors contributed to data collection, reviewed and approved the manuscript, and shared final responsibility for the decision to submit.
Conflict-of-interest disclosure: M.K. reports grants and other from AbbVie, grants and other from Genentech, grants and other from F. Hoffman La-Roche, grants and other from Stemline Therapeutics, other from Amgen, grants and other from Forty-Seven, other from Kisoji, grants from Eli Lilly, grants from Cellectis, grants from Calithera, grants from Ablynx, grants from Agios, grants from Ascentage, grants from Astra Zeneca, other from Reata Pharmaceutical, grants from Rafael Pharmaceutical, grants from Sanofi, outside the submitted work, and a patent US 7 795 305 B2 CDDO-compounds and combination therapies with royalties paid to Reata Pharm., a patent Combination Therapy with a mutant IDH1 inhibitor and a BCL-2 licensed to Eli Lilly, and a patent 62/993 166 combination of a MCL-1 inhibitor and midostaurin, uses and pharmaceutical compositions thereof pending to Novartis. E.J. reports research grants and consultancy from Abbvie, Adaptive Biotechnology, Amgen, BMS, Pfizer, and Takeda. N.D. reports research funding from Daiichi Sankyo, Bristol-Myers Squibb, Pfizer, Karyopharm, Sevier, Genentech, Astellas, Abbvie, Genentech, Novimmune, Amgen, Trovagene, Gilead, FATE Therapeutics, Trillium, Hanmi, Newave, Glycomimetics, and ImmunoGen; he has served in a consulting or advisory role for Daiichi Sankyo, Bristol-Myers Squibb, Pfizer, Novartis, Celgene, AbbVie, Genentech, Servier, Trillium, Syndax, Trovagene, Astellas, Gilead, STAR Therapeutics, KITE, and Agios. M. Andreeff has received grant or research support from Daiichi-Sankyo, Breast Cancer Research Foundation, ONO Pharmaceuticals, Karyopharm, Amgen, AstraZeneca, and Oxford Biomedica UK; paid consultancy from Daiichi-Sankyo, Aptose, Senti-Bio, Glycomimetics, Syndax, Medicxi; membership on advisory committees or review panels, board membership in Cancer UK, Leukemia & Lymphoma Society, German Research Council, NCI-RDCRN (Rare Disease Clinical Network), CLL Foundation, Novartis; and ownership interest (eg, stocks, stock options) in Reata, Aptose, Eutropics, SentiBio, Chimerix, and Oncolyze. S.M., M.C., and J.Z. are employed by and have equity ownership in IsoPlexis.
Correspondence: Naval Daver, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 0428, Houston, TX 77030; e-mail: ndaver@mdanderson.org.

