

Key Points
First report of an FLT3-targeted therapy added to nonintensive chemotherapy that has improved survival in older FLT3-ITD patients with AML.
Quizartinib is well tolerated, improves response and survival in older FLT3-ITD AML patients and merits consideration in future therapies. Now amended as text above.

Abstract
Survival for older patients with acute myeloid leukemia (AML) unsuitable for intensive chemotherapy is unsatisfactory. Standard nonintensive therapies have low response rates and only extend life by a few months. Quizartinib is an oral Fms-like tyrosine kinase 3 (FLT3) inhibitor with reported activity in wild-type patients. As part of the AML LI trial, we undertook a randomized evaluation of low-dose ara-C (LDAC) with or without quizartinib in patients not fit for intensive chemotherapy. Overall, survival was not improved (202 patients), but in the 27 FLT3-ITD patients, the addition of quizartinib to LDAC improved response (P = .05) with complete remission/complete remission with incomplete haematological recovery for quizartinib + LDAC in 5/13 (38%) vs 0/14 (0%) in patients receiving LDAC alone. Overall survival (OS) in these FLT3-ITD+ patients was also significantly improved at 2 years for quizartinib + LDAC (hazard ratio 0.36; 95% confidence intervals: 0.16, 0.85, P = .04). Median OS was 13.7 months compared with 4.2 months with LDAC alone. This is the first report of an FLT3-targeted therapy added to standard nonintensive chemotherapy that has improved survival in this population. Quizartinib merits consideration for future triplet-based treatment approaches. This trial was registered at www.clinicaltrials.gov as ISRCTN #ISRCTN40571019 and EUDRACT @2011-000749-19.
Introduction
Among patients with acute myeloid leukemia (AML) over the age of 60, a considerable number are not considered suitable for intensive remission induction chemotherapy. Survival in these patients is poor.1,2 The possibility of combination therapy with additional agents represents an attractive option. Quizartinib is an orally administered second-generation class III receptor tyrosine kinase inhibitor with potent and highly efficacious inhibitory activity against Fms-like tyrosine kinase 3 (FLT3) in vitro and in vivo.3 It binds the FLT3 receptor in the inactive conformation in a region adjacent to the ATP-binding domain, preventing activity of internal tandem duplication (ITD) but does not target tyrosine kinase domain (TKD) mutations.4 Early phase trials of quizartinib as monotherapy demonstrated acceptable toxicity, and potential activity in AML.4 Activity in wild-type FLT3 patients was postulated to be due to KIT, PDGFRa/b, RET, CSF1R/FMS activity in addition to FLT3.3 Although early development identified QTc prolongation as a potential adverse effect, these early evaluations were at doses of up to 300 mg.5 Subsequent studies at 60 mg have confirmed the activity and tolerability such that drug approval has been achieved in Japan.6
To assess the efficacy of quizartinib in older patients with AML considered unsuitable for intensive therapy, we incorporated the addition of quizartinib to low-dose ara-C (LDAC) vs LDAC alone in a “pick-a-winner” design.7 This design allows several treatments to be assessed simultaneously in a randomized fashion, with the aim of doubling 2-year survival from 11% to 22% (hazard ratio [HR] 0.69), with interim assessments after 50 and 100 patients per arm are recruited. At the time of trial design, a response rate of 36% had been reported in relapsed/refractory patients with AML without FLT3-ITD, and therefore, the plan was to enroll irrespective of FLT3 mutation status.5,8
Methods
Initially, quizartinib was given orally at 90 mg once daily for 21 consecutive days as 1 cycle of treatment; after initial analysis, and emergent data from other studies, the dose was amended to 60 mg once daily. LDAC was given at 20 mg twice daily subcutaneously on days 1 to 10 of each course, with courses occurring at 4- to 6-week intervals. To enter the randomization, patients needed to fulfill specific cardiac criteria (no significant ischemic heart disease, heart failure, prolonged QTc, or second/third-degree heart block). Electrolyte levels of potassium, magnesium, and calcium had to be within the normal range. Medications associated with QT/QTc prolongation and strong CYP3A4 inhibitors were not routine, although where considered standard care were allowed with dose reduction. Toxicities were recorded using Common Terminology Criteria for Adverse Events, version 3. The protocol defined complete remission (CR) as a normocellular bone marrow containing <5% leukemic blasts with evidence of normal maturation, neutrophil recovery to ≥1.0 × 109/L and platelets to ≥100 × 109/L. Patients who achieved CR according to the protocol, but without evidence of adequate count recovery are denoted here as complete remission with incomplete haematological recovery, and patients were required to be platelet transfusion independent, indicating sufficient time for marrow regeneration. FLT3-ITD detection was performed by reverse transcription-polymerase chain reaction and genomic DNA polymerase chain reaction using previously described primers,9 forward primer fluorescein amidites labeled, followed by fragment size analysis on the Applied Biosystems Genetic Analyzer 3130. Patients were considered to be FLT3-ITD+ with allelic ratio (AR) of at least 5%. The AR was derived using the European leukaemia net standard (the ratio of FLT3-ITD:WT calculated from the area under the curve(s) from the genetic analyzer). The AML Less Intensive Program (LI-1 Trial) has been approved by the Research Ethics Committee Wales. The study was conducted in accordance with the Declaration of Helsinki.
Results and discussion
Between July 2012 and May 2019, 202 patients from Denmark (1%), New Zealand (11%), and the United Kingdom (88%) were randomized. Median age was 77 years (range, 60 to 89). Overall, 64% were male; 63% had de novo AML; 25% had secondary AML; and 11% had high-risk myelodysplastic syndrome; 2% had favorable, 66% had intermediate, 27% had adverse, and 5% had unknown cytogenetics.10 An FLT3-ITD mutation was identified in 27 patients (16%), with a 5% cutoff for AR, and an FLT3-TKD mutation in 6 patients (3%); these results were not reported to investigators. Results here are based on median follow-up of 13.1 months. A median of 2 courses of therapy was delivered in either arm (mean, 2.7 for the LDAC arm, 3.0 for the LDAC+ quizartinib arm; range for both: 1 to 12). Fifty-two patients received the 90-mg dose; subsequently, 49 had the amended 60-mg dose.
In the trial population, overall response (CR/CRi) was achieved in 26 of 202 patients (13%), (LDAC+ quizartinib 16%, LDAC 10%, odds ratio 1.81; 95% confidence intervals, 0.73, 4.50; P = .199). Thirty-day mortality appeared to be no different (10% vs 15%; HR 0.71 (0.32, 1.60); P = .415); 2-year survival showed no significant difference (2-year overall survival [OS] 11% vs 17%, HR 0.87 (0.64, 1.19), 0.381). Median OS time was 5.5 vs 3.8 months (HR 0.89 (0.66, 1.21), P = .46). The response rate was unaltered by the dose reduction (13% vs 18%; 90 mg vs 60 mg, respectively).
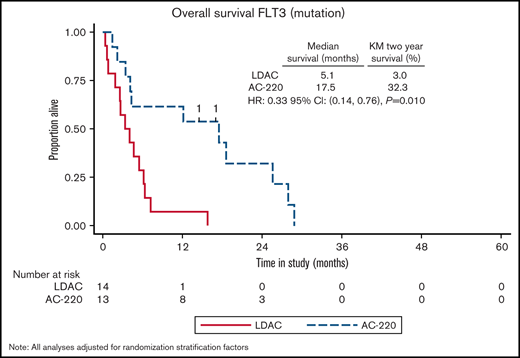
The initial data monitoring committee meeting reviewed the entire cohort; additional data from other studies informed the planned subgroup analyses stratified by FLT3-ITD. This identified a benefit from quizartinib in response (P = .02, P interaction .055) with CR/CRi for LDAC+ quizartinib in 5 of 13 patients (38%) and for LDAC alone in 0 of 14 patients (0%). The FLT3-ITD AR ranged from 0.03 to 2.03 (n = 25) with a median of 0.36 (interquartile range 0.36) and mean of 0.45 (standard deviation 0.42). Response did not correlate with FLT3-ITD AR (P = .9). Four patients had FLT3-ITD clones below the 5% cutoff, none of whom responded to AC220. Survival improved with the addition of quizartinib with a median OS of 13.7 months compared with 4.2 months with LDAC alone. OS at 2 years was also significantly better for recipients of LDAC+ quizartinib 26% vs 2% for LDAC alone (HR 0.36 [0.16, 0.85]; P = .024, P interaction .04; Figures 1 and 2).

The cause of death for most patients was resistant/recurrent disease: 59 (64%) in the LDAC+ quizartinib arm vs 59 (66%) in the LDAC-alone arm. Relapses occurred in 17 patients, which were too few to reliably determine relapse-free survival.
Quizartinib was associated with mostly grade 1 or 2 cardiac toxicities during the first course. Grade 3/4 events occurred in 8.7% of recipients of LDAC+ quizartinib vs 2.2% of LDAC-only treated patients. Examples included QTc prolongation (4 cases) and cardiac failure (1 case) and only occurred in the 90-mg group. Although there was 1 case of ventricular fibrillation in the 60-mg group, it was associated with severe sepsis and hypocalemia. In the quizartinib arm, there was no torsades de pointes or cardiac deaths, and reducing the dose had no impact on the response rates.
FLT3-ITD is a common driver mutation that presents with a high leukemic burden and confers a poor prognosis in patients with AML. These disease characteristics, response to therapy, and survival have been well described in younger patients with AML11 ; increasingly, these observations have been reproduced in older patients where survival appears especially poor in those treated with nonintensive chemotherapy.12,13
In parallel to the LI-1 trial, interim results of an uncontrolled physician’s choice study of quizartinib in combination with either azacitidine or LDAC have been reported in older patients with either newly diagnosed or relapsed FLT3-ITD+ AML also reported, encouraging activity.14
The data presented here are the first report of a randomized evaluation of a genomic targeted therapy being combined with nonintensive chemotherapy that has demonstrated an improvement in response and survival when compared with standard therapy alone. An especially low level of response to LDAC is well described in patients with AML with adverse karyotype,2 and here we report comparable findings for the FLT3-ITD subgroup. Although nontoxic and easy to administer, the specific contribution that LDAC provides to future combinatorial approaches is at best unclear as the response rate and survival from the LDAC+ quizartinib arm may be in keeping with what would be expected from quizartinib alone.
FLT3 inhibitors such as gilteritinib with azacitidine have well-defined in vitro activity. Furthermore, the combination of gilteritinib with azacitidine in murine AML xenograft models with FLT3-ITD demonstrated synergy over either treatment alone.15 Despite this, the randomized evaluation of the addition of gilteritinib to azacitidine has recently halted recruitment, as it did not meet its primary endpoint of OS at a planned interim analysis (LACEWING; #NCT02752035).16
Other FLT3 inhibitors assessed in a similar population include the combination of sorafenib with azacitidine17 with clinically meaningful response rates of 78%; this appeared well tolerated in small numbers of patients but has yet to be evaluated in a randomized study. Subsequent clinical focus is increasingly on second-generation, more potent, FLT3 inhibitors, which also target the TKD mutations, which are a frequent mechanism for the emergence of resistance.18
The recently published results for partnering azacitidine or LDAC with venetoclax have established a new standard of care.19,20 Understandably, there were only small numbers of FLT3-ITD patients in these studies, although response rates appear lower and less durable compared with the cohort. Primary and adaptive resistance to venetoclax-based combinations is most commonly characterized by the acquisition or enrichment of clones with activating signaling pathways, such as FLT3 or RAS.21 These observations have led to the global interest in genomically driven doublet22 and triplet23 combinations for future evaluation in which targeted therapies are combined to improve the durability of response by disrupting these emergent resistant clones. Therefore, the observation of efficacy and tolerability of quizartinib in older patients with AML as reported in the LI-1 trial should inform the rational development of such triplet combinations for future studies.
Acknowledgments
The authors are grateful to Daiichi Sankyo for providing drugs and support.
The authors are grateful to the National Institute for Health Research for supporting local trial delivery, Blood Cancer UK for research support, the Haematology Clinical Trials Unit and Centre for Trials Research, and Cardiff University for managing the trial.
Authorship
Contribution: M.D. was the chief investigator, reviewed the data, and wrote the manuscript; A.K.B. designed the trial, wrote the protocol, and was chief investigator until Q3 2014; R.K.H. designed the trial and wrote the protocol; I.F.T. supervised the data collection and reviewed the data; C.A. analyzed the data; L.U. supervised the data collection and reviewed the data; R.R., C.H., and P.M. were major recruiters; N.R. designed the trial and reviewed the data; M.C., co- chief investigator, S.K., and R.E.C. reviewed the data; and A.G. performed molecular analysis. All authors reviewed the manuscript.
Conflict-of-interest disclosure: A.K.B. was an employee of CTI Biopharma 2015-2017. R.E.C. has received research funding and honoraria from Novartis and Bristol Myers Squibb, and honoraria from Pfizer, Jazz Pharmaceuticals, and Abbvie in the past 3 years. M.C. has received research funding from Novartis, Bristol Myers Squibb, Cyclacel, and Takeda/Incyte, is or has been an advisory board member for Bristol Myers Squibb, Novartis, Incyte, Daiichi Sankyo, Jazz, and Pfizer and has received honoraria from Astellas, Bristol Myers Squibb, Novartis, Incyte, Pfizer, and Gilead. The remaining authors declare no competing financial interests.
Correspondence: Mike Dennis, The Christie NHS FT, Manchester, M20 4BX United Kingdom; e-mail: mike.dennis1@nhs.net.

