

Key Points
In vivo HSPC transduction with vectorized base editors efficiently installs target base conversions and reactivates γ-globin expression.
In vivo base editing is safe and generates significantly lower levels of indels and intergenic deletions than the CRISPR system.

Abstract
Base editors are capable of installing precise genomic alterations without creating double-strand DNA breaks. In this study, we targeted critical motifs regulating γ-globin reactivation with base editors delivered via HDAd5/35++ vectors. Through optimized design, we successfully produced a panel of cytidine and adenine base editor (ABE) vectors targeting the erythroid BCL11A enhancer or recreating naturally occurring hereditary persistence of fetal hemoglobin (HPFH) mutations in the HBG1/2 promoter. All 5 tested vectors efficiently installed target base conversion and led to γ-globin reactivation in human erythroid progenitor cells. We observed ~23% γ-globin protein production over β-globin, when using an ABE vector (HDAd-ABE-sgHBG-2) specific to the –113A>G HPFH mutation. In a β-YAC mouse model, in vivo hematopoietic progenitor/stem cell (HSPC) transduction with HDAd-ABE-sgHBG-2 followed by in vivo selection resulted in >40% γ-globin+ erythrocytes in the peripheral blood. This result corresponded to 21% γ-globin production over human β-globin. The average –113A>G conversion in total bone marrow cells was 20%. No alterations in hematological parameters, erythropoiesis, and bone marrow cellular composition were observed after treatment. No detectable editing was found at top-scoring, off-target genomic sites. Bone marrow lineage–negative cells from primary mice were capable of reconstituting secondary transplant-recipient mice with stable γ-globin expression. Importantly, the advantage of base editing over CRISPR/Cas9 was reflected by the markedly lower rates of intergenic HBG1/2 deletion and the absence of detectable toxicity in human CD34+ cells. Our observations suggest that HDAd-vectorized base editors represent a promising strategy for precise in vivo genome engineering for the treatment of β-hemoglobinopathies.
Introduction
Most genome engineering strategies based on nucleases such as CRISPR/Cas9 rely on double-strand DNA breaks (DSBs), which may cause critical side effects in host cells by generating unwanted large genomic deletions and p53-dependent DNA damage responses.1-3 Base editors (BEs) can generate precise nucleotide substitutions at targeted genomic loci without creating DSBs. They comprise a catalytically disabled nuclease, such as Cas9 nickase (nCas9), that is incapable of making DSBs and is fused to a nucleobase deaminase enzyme and, in some cases, a DNA glycosylase inhibitor. Currently, there are 2 major categories, cytidine base editors (CBEs) and adenine base editors (ABEs), which catalyze C>T and A>G transitions, respectively, in a narrow targetable window (usually ∼5 bp) dictated by a single guide RNA (sgRNA) coupled with the nCas9.4-6 The key difference between CBEs and ABEs is in the deaminase domain; CBEs contain a cytidine deaminase (eg, APOBEC1) whereas ABEs use laboratory-evolved TadA deoxyadenosine deaminases. Multiple groups have reported efficient base editing in a variety of eukaryotic cells.7-11 It is thought that ∼60% of all known pathogenic single-nucleotide polymorphisms in humans can be reversed by current BEs.12
After birth, fetal hemoglobin (HbF or α2/γ2) is replaced by adult hemoglobin, with HbA (α2/β2) being the most common form in adults. β-Hemoglobinopathies are a common group of genetic disorders with absent or deficient production of β-globin, mainly β-thalassemia and sickle cell disease (SCD). Depending on the specific genetic defects, β-thalassemia and patients with SCD exhibit various disease severities. Most patients with β-thalassemia major (β0) and SCD have lifelong acute and chronic complications.13,14 However, in some adult patients with high levels of HbF, the disease symptoms are markedly milder. This phenomenon of hereditary persistence of fetal hemoglobin (HPFH) demonstrates a strong protective effect of HbF and provides a rationale for reactivation of γ-globin as a gene therapy strategy for patients with β-globin disorders.
Several HPFH mutations have been reported (reviewed by Orkin et al15 and Wienert et al16 ). In the promoters of the γ-globin–encoding HBG1 and HBG2 genes, there are 3 major clusters of HPFH-related single-nucleotide polymorphisms located at the –115-, –175-, and –200-bp sites. Introduction of HPFH mutations at these sites can disrupt the binding of HBG1/2 repressor proteins (eg, BCL11A and ZBTB7A) or create gain-of-function binding sites for activators (eg, TAL1 and KLF1), leading to derepressed γ-globin expression.17,18 γ-Globin reactivation can also be achieved by modulating the expression of the HBG1/2 gene repressors, such as BCL11A.19 Although direct BCL11A knockout is not possible because of its developmentally indispensable roles, partial downregulation of BCL11A by editing its erythroid-specific enhancer allows for efficient γ-globin induction without significant side effects.20,21 This result has been demonstrated by ex vivo transfection of human CD34+ hematopoietic stem/progenitor cells (HSPCs) using A3A(N57Q)-BE3 CBEs that target a critical site in the BCL11A enhancer.9
We have recently established a simplified gene therapy approach involving in vivo transduction of mobilized HSPCs. The central idea of this in vivo approach is to mobilize HSPCs from the bone marrow, and, although they circulate at high numbers in the periphery, transduce them with an IV injected HSPC-tropic, helper-dependent, adenovirus HDAd5/35++ gene transfer vector system. Transduced cells return to the bone marrow where they persist for the long term. For gene addition strategies, random transgene integration in HSPCs can be achieved by a Sleeping Beauty transposase (SB100×) system.22 This system consists of 2 HDAd5/35++ vectors. The first is a transposon vector where the transgene cassette is flanked by inverted repeats (IRs), which are recognized by the SB100× transposase and frt sites that allow for circularization of the transgene cassette in the presence of Flpe recombinase. The second vector, HDAd-SB, supplies Flpe recombinase, and SB100× in trans to mediate integration of the expression cassette into TA dinucleotides of the genomic DNA.23 To achieve 80% to 100% transgene marker levels in peripheral blood cells, an in vivo selection system is currently used. In this system, in addition to therapeutic genes, a P140K mutant of the human O-6-methylguanine-DNA methyltransferase (mgmt) gene is expressed from the HDAd5/35++ vector. The mgmtP140K gene confers drug resistance and the selective expansion of gene-modified cells upon selection with the mgmt inhibitor O6-benzylguanine (O6BG) and carmustine (BCNU). We demonstrated the safety and efficacy of our approach in mouse models for thalassemia intermedia, SCD, and hemophilia A, where we achieved phenotypic corrections.24-27 Furthermore, our ongoing studies in nonhuman primates have confirmed the effectiveness and safety of in vivo HSPC transduction.28
For genome editing, only short-term expression of BEs is necessary. In this study, we used the SB100× system to achieve this expression. In HDAd-BE vectors, the BE expression cassette was placed into the HDAd5/35++ vector genome outside an frt/IR-flanked mgmt/GFP unit. Coinfection with an HDAd-SB vector results in Flpe/SB100×-mediated integration of the mgmt/GFP cassette, whereas the remaining parts of the HDAd-BE genome, including the BE expression unit, are rapidly degraded.29 Furthermore, the system allows for mgmtP140K-based in vivo selection of transduced and genome-edited HSPCs.
In this study, using an optimized design, we successfully generated a panel of HDAd-BE vectors targeting the erythroid BCL11A enhancer or the HBG1/2 promoter. In CD46/β-YAC transgenic mice, we demonstrated that in vivo HSPC base editing with an HDAd-ABE vector created HPFH mutations and led to efficient γ-globin induction.
Methods
Animal studies
All experiments involving animals were conducted in accordance with the institutional guidelines set forth by the University of Washington. The studies were approved by the University’s Institutional Animal Care and Use Committee (protocol no. 3108-01). C57BL/6J-based transgenic mice that contain the human CD46 genomic locus and provide CD46 expression at a level and in a pattern similar to that expressed in humans (hCD46+/+ mice) have been described earlier.30 Transgenic mice carrying the wild-type 248-kb β-globin locus yeast artificial chromosome (β-YAC) were used.31 β-YAC mice were crossed with human CD46+/+ mice to obtain β-YAC+/−/CD46+/+ mice for in vivo HSPC transduction studies. HSPC mobilization and in vivo transduction, in vivo selection, secondary bone marrow transplantation, and transplantation of human CD34+ cells are described in the supplemental Information.
Other methods
Generation of HDAd vectors, transfection of cell lines, HUDEP-2 cell and erythroid differentiation, CD34+ cell culture, tissue and blood analyses, colony-forming unit (CFU) assay, T7EI mismatch nuclease assay, measurement of base conversion by Sanger sequencing and next-generation sequencing (NGS), flow cytometry, globin high-performance liquid chromatography (HPLC), real-time reverse transcription-polymerase chain reaction (PCR), measurement of vector copy number, and statistical analyses are described in the supplemental Materials and Methods. The oligonucleotides used for cloning are shown in supplemental Table 4.
Results
Selection of BEs and guide RNAs
We compared the editing activity of multiple versions of CBEs including BE4,32 AncBE4max,33 BE3RA, and FNLS.34 The efficacy of the CBE was measured by using T7 endonuclease-1, an enzyme that cleaves structural deformities in DNA heteroduplexes created by CRISPR/Cas9 or BEs (T7EI mismatch assay). Although the editing efficiency in 293FT cells was comparable for all 4 CBEs in the range of 12.7% to 15.5% (supplemental Figure 1A), the AncBE4max system was superior in K562 erythroleukemia cells (supplemental Figure 1B). Therefore, we selected the AncBE4max platform for our studies. For ABE, we chose the ABEmax system developed and optimized by David Liu’s group.33 The xCas9(3.7)-BE4 and xCas9(3.7)-ABE(7.10) editors with broad protospacer adjacent motif (PAM) compatibility (NGN motif) were also used to obtain more guide sequence options.35
For our HDAd-delivered base editing approach, we screened a series of sgRNAs (1) specific to the functionally critical GATAA motif in the +58 region of the erythroid BCL11A enhancer (sgBCL-1 to -6) or (2) capable of recreating various HPFH mutations in the HBG1/2 promoter at clusters –115, –175, and –200 (sgHBG-1 to -6). The sgRNAs were designed to have the optimal targetable window of BEs set at positions 4 to 8 of the protospacer (counting the 5′-end first base as position 1). The sequences and their specific target motifs and bases are shown in Figure 1A. The guide sequences, cloned into the ABE and CBE vectors, were tested in the erythroid progenitor HUDEP-2 cell line,36 for their potency in reactivating γ-globin expression. At day 4 after transfection, HUDEP-2 cells were subjected to erythroid differentiation. All 12 ABE and CBE variants led to significant γ-globin expression compared with the nontransduced controls. A negative control (CBE) that targeted CCR5, but not hemoglobin-related genes, did not reactivate γ-globin (Figure 1B). An ABE vector (sgHBG-2; mapping to –113A site) resulted in 41% γ-globin+ cells at day 6 after erythroid differentiation. A previously described HDAd5/35++ CRISPR/Cas9 vector targeting the BCL11A binding site in the HBG promoter was used as a positive control and generated 84% of the γ-globin+ cells.27 Using Sanger sequencing to determine base editing efficiency, we detected up to 15% conversion of sgBCL-1 to -4 and sgHBG-1 to -4. For ABEs and CBEs with xCas9 (sgBCL-5 to -6, sgHBG-5 to -6), the conversion level was ∼5% (Figure 1C). Accordingly, we chose sgBCL-1 (CBE), sgHBG-1 (CBE), sgHBG-2 (ABE), and sgHBG-4 (ABE) for HDAd vector delivery in consideration of their activity and diversity of target sites. Furthermore, a negative control (sgNeg/CBE) and a combination of sgHBG-1 and sgBCL-1 (CBE) were selected for HDAd production. The hypothesis beyond the multiplex vector was that dual targeting would have an additive effect on γ-globin reactivation.
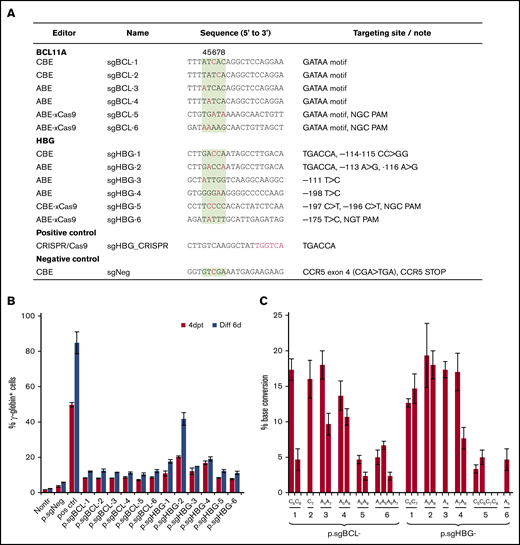
Screening of guide sequences. (A) Guide sequences with editor type and targeting site information. Red, targeted bases in critical motifs; green background, optimal targetable window (also indicated by the numbers above the column). A CRISPR/Cas9 plasmid targeting the TGACCA motif in the HBG1/2 promoter was used as a positive control. A CBE plasmid targeting the CCR5 coding region was included as a negative control (sgNeg). (B) HUDEP-2 cells were transfected with plasmids expressing guide sequences and the BEs listed in panel A. For example, p.sgBCL-1 indicates the plasmid-expressing guide sequence sgBCL-1. γ-Globin expression was measured by flow cytometry 4 days after transfection (4dpt) and 6 days after in vitro erythroid differentiation (Diff 6d). Nontr, nontransduced. (C) The percentage of base conversion detected at 4dpt by Sanger sequencing. Data are the mean ± SD of 3 biological replicates.
Screening of guide sequences. (A) Guide sequences with editor type and targeting site information. Red, targeted bases in critical motifs; green background, optimal targetable window (also indicated by the numbers above the column). A CRISPR/Cas9 plasmid targeting the TGACCA motif in the HBG1/2 promoter was used as a positive control. A CBE plasmid targeting the CCR5 coding region was included as a negative control (sgNeg). (B) HUDEP-2 cells were transfected with plasmids expressing guide sequences and the BEs listed in panel A. For example, p.sgBCL-1 indicates the plasmid-expressing guide sequence sgBCL-1. γ-Globin expression was measured by flow cytometry 4 days after transfection (4dpt) and 6 days after in vitro erythroid differentiation (Diff 6d). Nontr, nontransduced. (C) The percentage of base conversion detected at 4dpt by Sanger sequencing. Data are the mean ± SD of 3 biological replicates.
Generation of helper-dependent adenovirus vectors expressing BEs
The total length of BE genes plus regulatory elements exceeds 8 kb and is therefore beyond the payload capacity of lentivirus or adeno-associated virus vectors.37 HDAd5/35++ vectors that can package genomes of ∼32 kb can address this problem. In our first attempt, the BE enzyme (rAPOBEC1-nCas9-2×UGI for CBE; 2×TadA-nCas9 for ABE) driven by an EF1α pol II promoter and the sgRNA driven by a human U6 pol III promoter were cloned into the HDAd plasmid. An mgmt/GFP cassette flanked by frt and transposon IRs was also cloned into the vector to mediate selection of transduced cells by O6BG/BCNU (Figure 2A). Notably, the BE components were placed outside the SB100× transposon, only allowing for their transient expression, while maintaining integrated expression of the mgmt/GFP cassette upon codelivery with an HDAd-SB vector expressing SB100× transposase/flippase.38 Although the virus yield was relatively low (1 × 1012 viral particles per 2-L spinner flask with 6 × 108 HDAd producer cells), we were able to generate all 4 CBE vectors (supplemental Figure 2A), in contrast to HDAd-CRISPR/Cas9 vectors, which cannot be produced without using micro-RNA (mi-RNA)–based mechanisms that downregulate Cas9 expression in HDAd producer cells.39 Our results suggest that DSB-free BE systems may be less toxic to the HDAd producer cells than CRISPR/Cas9. For the ABE vectors, the virus genome appeared rearranged, and no distinct HDAd virus band was observed after ultracentrifugation in CsCl gradients. Because the major difference between ABE and CBE vectors was the deaminase domain, it was likely that the two 594 bp TadA-32aa repeats in ABE vectors were the elements causing recombination and rearrangements within the HDAd genome. To address this problem, the following modifications to the original version of the ABE vectors were made: (1) the sequence repetitiveness between the 2 TadA-32aa repeats was reduced by use of alternative codons (supplemental Figure 2B); and (2) a PGK promoter was used to drive the BE enzyme expression. Although it is highly active in HSPCs,40 the PGK promoter exhibits lower activity in 116 producer cells than the EF1α promoter,41 thereby reducing potential TadA-associated adverse effects; (3) an miR183/218-based gene regulation system was used to further suppress BE expression in 116 cells, while allowing for it in HSPCs39 (Figure 2A). This second version of ABE constructs with optimized design led to the successful production of 2 HDAd-ABE viruses with an average yield of 3.3 × 1012 viral particles (vp) per spinner flask, which is within the normal yield range (supplemental Figure 2A).
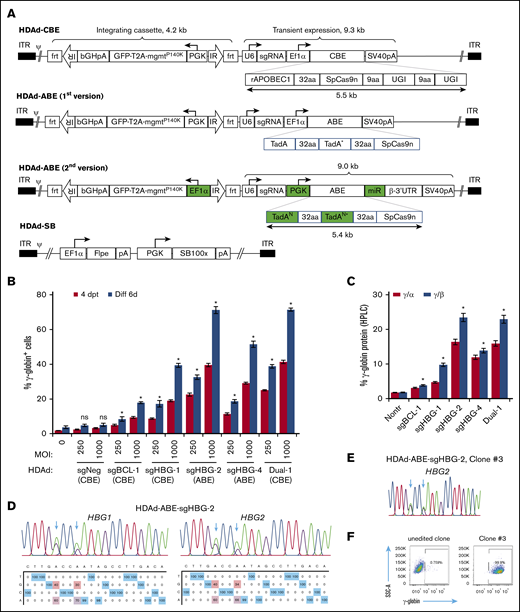
Construction and validation of HDAd-BE vectors. (A) Diagrams of HDAd vectors. The overall structure of HDAd-CBE/ABE vectors contains an mgmtP140K/GFP transposon (4.2 kb) flanked by 2 frt-IRs and a BE cassette (∼9 kb). The transposon allows for integrated expression when codelivered with HDAd-SB expressing SB100× transposase and flippase (Flpe). The BE cassette was placed outside of the transposon for transient expression. The first version of HDAd-ABE vectors was not producible. The second version of HDAd-ABE vector design contains 2 codon-optimized TadAN repeats to reduce sequence repetitiveness (N, new; *, the catalytic repeat). An mi-RNA–responsive element (miR) was embedded in the 3′ human β-globin untranslated region to minimize toxicity to producer cells by specifically downregulating ABE expression in 116 cells. 32aa or 9aa, linker with 32 or 9 amino acids; ψ, packaging signal; bGHpA, bovine growth hormone polyadenylation sequence; ITR, inverted terminal repeat; PGK, human PGK promoter; rAPOBEC1, cytidine deaminase enzyme; SpCas9n, SpCas9 nickase; SV40pA, simian virus 40 polyadenylation signal; T2A, a self-cleaving 2A peptide; TadA, adenosine deaminase; U6, human U6 promoter; and UGI, uracil glycosylase inhibitor. (B) γ-Globin induction in HUDEP-2 cells after transduction with HDAd vectors. Cells were transduced with various vectors at MOIs of 250 and 1000 vp per cell. The γ-globin expression was measured by flow cytometry 4 days after transfection (4dpt) and 6 days after in vitro erythroid differentiation (Diff 6d). A CBE vector targeting the CCR5 coding region was included as a negative control (sgNeg). Data shown are the mean ± SD of 3 biological replicates. (C) Percentage of γ-globin expression over α- or β-globin measured by HPLC at day 6 after differentiation. MOI of 1000 vp per cell. Data shown are the mean ± SD of 3 biological replicates. (D) Representative target base conversion by HDAd-ABE-sgHBG-2. HBG1 or HBG2 genomic segments encompassing the targeting bases were amplified and subjected to Sanger sequencing. Data were analyzed by EditR1.0.9. The arrows indicate targeted bases. The percentage of conversions are shown below the chromatograms. (E-F) A representative clone (#3) derived from HUDEP-2 cells transduced with HDAd-ABE-sgHBG-2. Monoallelic –113 and –116A>G base conversions were detected in the HBG1 promoter (E), resulting in 100% γ-globin+ cells detected by flow cytometry (F). *P < .05; ns, not significant (compared with nontransduced samples).
Construction and validation of HDAd-BE vectors. (A) Diagrams of HDAd vectors. The overall structure of HDAd-CBE/ABE vectors contains an mgmtP140K/GFP transposon (4.2 kb) flanked by 2 frt-IRs and a BE cassette (∼9 kb). The transposon allows for integrated expression when codelivered with HDAd-SB expressing SB100× transposase and flippase (Flpe). The BE cassette was placed outside of the transposon for transient expression. The first version of HDAd-ABE vectors was not producible. The second version of HDAd-ABE vector design contains 2 codon-optimized TadAN repeats to reduce sequence repetitiveness (N, new; *, the catalytic repeat). An mi-RNA–responsive element (miR) was embedded in the 3′ human β-globin untranslated region to minimize toxicity to producer cells by specifically downregulating ABE expression in 116 cells. 32aa or 9aa, linker with 32 or 9 amino acids; ψ, packaging signal; bGHpA, bovine growth hormone polyadenylation sequence; ITR, inverted terminal repeat; PGK, human PGK promoter; rAPOBEC1, cytidine deaminase enzyme; SpCas9n, SpCas9 nickase; SV40pA, simian virus 40 polyadenylation signal; T2A, a self-cleaving 2A peptide; TadA, adenosine deaminase; U6, human U6 promoter; and UGI, uracil glycosylase inhibitor. (B) γ-Globin induction in HUDEP-2 cells after transduction with HDAd vectors. Cells were transduced with various vectors at MOIs of 250 and 1000 vp per cell. The γ-globin expression was measured by flow cytometry 4 days after transfection (4dpt) and 6 days after in vitro erythroid differentiation (Diff 6d). A CBE vector targeting the CCR5 coding region was included as a negative control (sgNeg). Data shown are the mean ± SD of 3 biological replicates. (C) Percentage of γ-globin expression over α- or β-globin measured by HPLC at day 6 after differentiation. MOI of 1000 vp per cell. Data shown are the mean ± SD of 3 biological replicates. (D) Representative target base conversion by HDAd-ABE-sgHBG-2. HBG1 or HBG2 genomic segments encompassing the targeting bases were amplified and subjected to Sanger sequencing. Data were analyzed by EditR1.0.9. The arrows indicate targeted bases. The percentage of conversions are shown below the chromatograms. (E-F) A representative clone (#3) derived from HUDEP-2 cells transduced with HDAd-ABE-sgHBG-2. Monoallelic –113 and –116A>G base conversions were detected in the HBG1 promoter (E), resulting in 100% γ-globin+ cells detected by flow cytometry (F). *P < .05; ns, not significant (compared with nontransduced samples).
Next, we tested the HDAd-BE vectors in HUDEP-2 cells. All 5 tested vectors efficiently installed target base conversions and led to vector-dose–dependent γ-globin reactivation (Figure 2; supplemental Figure 3). Consistent with the plasmid screening data in Figure 1, infection with HDAd-ABE-sgHBG-2 resulted in the highest level of γ-globin+ cells (71% at a multiplicity of infection [MOI] of 1000 vp per cell). Interestingly, although the sgBCL-1 and sgHBG-1 BE vectors alone resulted in 17% and 39% γ-globin+ cells, respectively, the dual-targeting vector that simultaneously expressed sgBCL-1 and sgHBG-1 BEs, generated 71% γ-globin+ cells (Figure 2B), indicating a synergistic effect. No significant γ-globin induction was measured for the negative control vector. The γ-globin protein levels measured by HPLC were consistent with the flow cytometry data; 23% γ-globin over β-globin was observed after transduction with 1000 vp per cell HDAd-ABE-sgHBG-2 (Figure 2C). At an MOI of 1000, the base conversion frequencies for the 4 sgRNAs were in the range of 25% to 51% (Figure 2D; supplemental Figure 3A). For HDAd-ABE-sgHBG-2, ∼40% and 34% A>G conversions were detected at positions 5 and 8, respectively (Figure 2D). The A8>G conversion created the –113A>G HPFH mutation (Figure 1A).42 In single-cell–derived clones, monoallelic edits at sites A5 and A8 in the HBG2 promoter alone conferred 100% γ-globin+ cells (Figure 2E-F), confirming the critical role of these sites in regulating γ-globin suppression. Similar results were obtained in clones derived from HDAd-CBE-sgHBG-1– and HDAd-ABE-sgHBG-4–infected HUDEP-2 cells. In clones transduced with HDAd-CBE-sgBCL-1, a biallelic G>A mutation in the GATAA motif led to 15% of the cells expressing γ-globin (supplemental Figure 3B-C). Collectively, these data demonstrate that HDAd-BE vectors specific to critical sites in the erythroid BCL11A enhancer or HBG1/2 promoter can efficiently reactivate γ-globin expression in HUDEP-2 cells.
Reactivation of γ-globin in β-YAC mice after in vivo HSPC transduction with HDAd vectors expressing BE
We used β-YAC mice that contain 248 kb of human DNA in their genome, including the complete 82-kb β-globin locus.43 The mice were crossed with human CD46 transgenic mice to allow for transduction with HDAd5/35++ vectors, which use CD46 as a receptor. The HDAd-ABE-sgHBG-2 vector was selected for in vivo studies based on its superiority in inducing γ-globin expression in HUDEP-2 cells. After mobilization with G-CSF/AMD3100, β-YAC/CD46 mice were IV injected with HDAd-ABE-sgHBG-2 and HDAd-SB vectors. Four weeks after transduction, the mice were subjected to 4 rounds of O6BG/BCNU treatment to selectively expand HSPCs with integrated mgmtP140K/GFP transgenes (Figure 3A). After selection, the GFP marker reached 60% in peripheral blood mononuclear cells (PBMCs; Figure 3B-C). Notably, γ-globin expression in peripheral red blood cells (RBCs) increased from ∼1% before transduction to an average of 43% (n = 9) at week 16 after transduction, demonstrating significant expansion of genome-edited cells (Figure 3D-E). The variation observed between different mice was probably caused by the bicistronic design of mgmt-2A-GFP, which could result in lower expression of MGMTP140K and could therefore have affected the efficacy of in vivo selection. γ-Globin+ cells resided in the RBC fraction (Ter-119+; Figure 3F). In RBC lysates at week 16, up to 21% human γ-globin protein over human β-globin was measured by HPLC (Figure 3G; supplemental Figure 4). γ-Globin mRNA expression levels were in agreement with the HPLC data (Figure 3H). In total bone marrow mononuclear cells at week 16, the average integrated vector copy number was 1.4 per cell (Figure 3I).
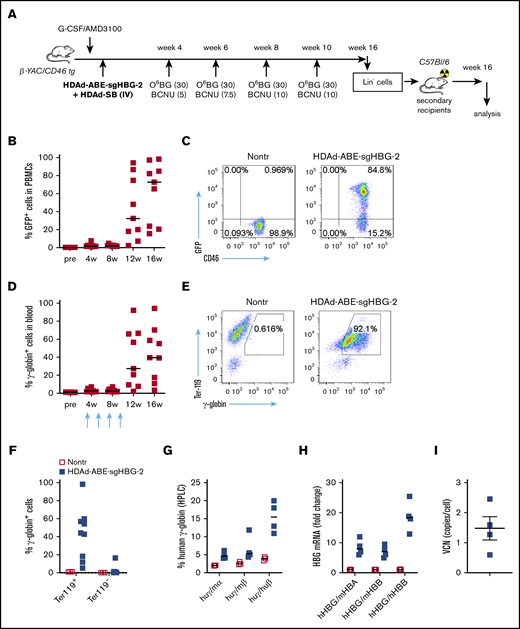
Reactivation of γ-globin in β-YAC mice after in vivo HSPC transduction and selection. (A) Experimental procedure. β-YAC/CD46 mice (n = 9) were mobilized by G-CSF/AMD3100 and in vivo transduced with HDAd-ABE-sgHBG-2+HDAd-SB. Four rounds of selection by O6BG/BCNU were performed at 4, 6, 8, and 10 weeks after transduction. The mice were killed at week 16. Lin− cells were isolated from bone marrow and IV injected into lethally irradiated C57BL/6J mice. The secondary transplanted mice were observed for another 16 weeks. (B) GFP markers in PBMCs at various time points after transduction. Each symbol represents 1 animal. (C) Flow cytometry showing GFP expression in PBMCs in 1 animal. (D) γ-Globin expression in peripheral RBCs measured by flow cytometry. (E) Flow cytometry charts showing γ-globin expression in total blood cells in a representative animal. (F) γ-Globin expression measured by flow cytometry in erythroid Ter-119+ and nonerythroid Ter-119– cells in blood. (G) Human γ-globin chain levels in RBC lysates measured by HPLC. Data are percentages of γ-globin chain levels over mouse α- or β-globin or human β-globin. (H) γ-Globin expression at mRNA level measured by reverse transcription-PCR. Data are expressed as fold change over mouse HBA or HBB or over human HBB mRNA. (I) Vector copy number (VCN) in total bone marrow cells. Primers specific to human mgmtP140K were used.
Reactivation of γ-globin in β-YAC mice after in vivo HSPC transduction and selection. (A) Experimental procedure. β-YAC/CD46 mice (n = 9) were mobilized by G-CSF/AMD3100 and in vivo transduced with HDAd-ABE-sgHBG-2+HDAd-SB. Four rounds of selection by O6BG/BCNU were performed at 4, 6, 8, and 10 weeks after transduction. The mice were killed at week 16. Lin− cells were isolated from bone marrow and IV injected into lethally irradiated C57BL/6J mice. The secondary transplanted mice were observed for another 16 weeks. (B) GFP markers in PBMCs at various time points after transduction. Each symbol represents 1 animal. (C) Flow cytometry showing GFP expression in PBMCs in 1 animal. (D) γ-Globin expression in peripheral RBCs measured by flow cytometry. (E) Flow cytometry charts showing γ-globin expression in total blood cells in a representative animal. (F) γ-Globin expression measured by flow cytometry in erythroid Ter-119+ and nonerythroid Ter-119– cells in blood. (G) Human γ-globin chain levels in RBC lysates measured by HPLC. Data are percentages of γ-globin chain levels over mouse α- or β-globin or human β-globin. (H) γ-Globin expression at mRNA level measured by reverse transcription-PCR. Data are expressed as fold change over mouse HBA or HBB or over human HBB mRNA. (I) Vector copy number (VCN) in total bone marrow cells. Primers specific to human mgmtP140K were used.
We performed Sanger sequencing and NGS to measure the frequency of base conversion in bone marrow cells. Results from the 2 approaches were comparable (supplemental Figure 5). The average A>G conversion at sites A5 and A8 was15% to 30% (Figure 4A-B). In the mouse with the highest γ-globin expression, ∼82% base conversion was achieved (Figure 4B). The base conversion at site A8 in the HBG1 region was slightly higher than that in the HBG2 region (26% vs 20%, on average). Notably, the percentage of conversion at site A5 tended to be slightly higher than that at A8 in both the HBG1 and HBG2 regions, although no significant difference was found (Figure 4B-C). The γ-globin expression levels were found to be directly correlated with the base-editing frequencies (Pearson test, R = 0.92; P < .001; Figure 4D). It has been shown that some BEs exhibit processive editing when multiple target bases are present in the protospacer. We detected minor editing with 3% and 4% A>G conversions at sites A9 and A11, respectively (Figure 4E). NGS results showed that base substitutions were predominantly A>G. Byproducts including A>C and A>T edits were <0.5% (Figure 4E). Importantly, the DSB-independent nature of BEs was reflected by a very low frequency (<0.5%) of NGS reads with insertions or deletions at the target sites (Figure 4F). In summary, these data demonstrate that in vivo HSPC transduction with HDAd-ABE-sgHBG-2 specific to the HBG1/2 promoter followed by selection leads to efficient target base conversion and γ-globin induction in β-YAC/CD46 mice.
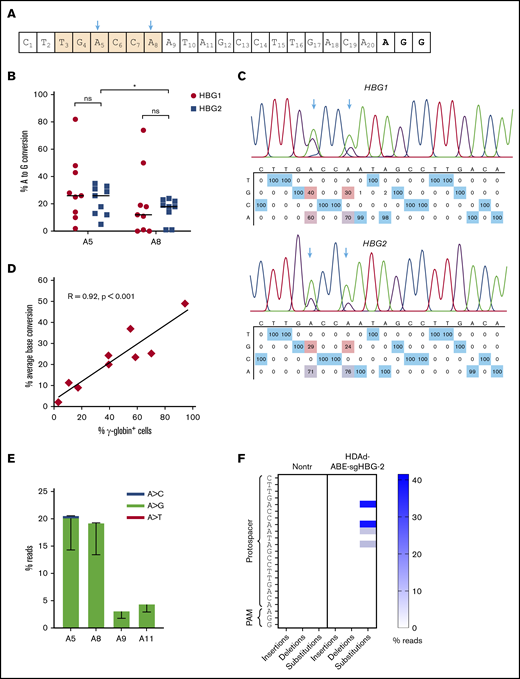
Target base conversion after in vivo HSPC transduction of β-YAC mice with HDAd-BE vectors. (A) sgHBG-2 guide sequence. The numbering starts from the 5′ end. Highlighted in orange is the TGACCA motif, a reported BCL11A binding site within the HBG1/2 promoters. The 2 adenines (A5 and A8) in the motif are indicated by arrows. (B) Percentage of target base conversion in total bone marrow mononuclear cells by Sanger sequencing. Both A5 and A8 in HBG1 and HBG2 promoter regions are shown. Each symbol represents 1 animal (n = 9). (C) Representative sequencing results showing target base conversion in HBG1 and HBG2 region of the #1369 mouse. (D) Correlation between average base conversions and γ-globin expression. The percentage of average base conversion in each animal was the average level at A5 and A8 in HBG1 and HBG2 promoter regions. Each symbol represents 1 animal (n = 9). A Person test was performed (R = 0.92; P < .001). (E) Substitution frequencies detected by NGS; n = 5). (F) Frequency of insertions, deletions, and substitutions in mouse #1369 measured by NGS and analyzed by CRISPResso2. *P < .05. ns, not significant.
Target base conversion after in vivo HSPC transduction of β-YAC mice with HDAd-BE vectors. (A) sgHBG-2 guide sequence. The numbering starts from the 5′ end. Highlighted in orange is the TGACCA motif, a reported BCL11A binding site within the HBG1/2 promoters. The 2 adenines (A5 and A8) in the motif are indicated by arrows. (B) Percentage of target base conversion in total bone marrow mononuclear cells by Sanger sequencing. Both A5 and A8 in HBG1 and HBG2 promoter regions are shown. Each symbol represents 1 animal (n = 9). (C) Representative sequencing results showing target base conversion in HBG1 and HBG2 region of the #1369 mouse. (D) Correlation between average base conversions and γ-globin expression. The percentage of average base conversion in each animal was the average level at A5 and A8 in HBG1 and HBG2 promoter regions. Each symbol represents 1 animal (n = 9). A Person test was performed (R = 0.92; P < .001). (E) Substitution frequencies detected by NGS; n = 5). (F) Frequency of insertions, deletions, and substitutions in mouse #1369 measured by NGS and analyzed by CRISPResso2. *P < .05. ns, not significant.
Safety profile and durable effect of in vivo HSPC base editing
At week 16 after in vivo transduction, the animals were euthanized, and tissue samples were subjected to hematological and histological analyses. Hematological parameters, including white blood cells (1 000/μL), RBCs (1 000 000/μL), Hb (g/dL), mean corpuscular volume (fL), mean corpuscular hemoglobin concentration (g/dL), RBC distribution (%), and platelets (1 000/μL), were similar to that of naive β-YAC/CD46 mice (Figure 5A). The percentage of reticulocytes in blood was unaffected (Figure 5B). No foci of extramedullary erythropoiesis were observed on spleen and liver sections (supplemental Figure 6). The cellular composition in PBMCs, spleen and bone marrow were indistinguishable from the composition in control mice (Figure 5C). Furthermore, compared with other previously reported HDAd5/35++ gene therapy vectors,25,27,44 HDAd-ABE-sgHBG-2 did not cause significant changes in body weight, behavior, and appearance after in vivo transduction and selection.
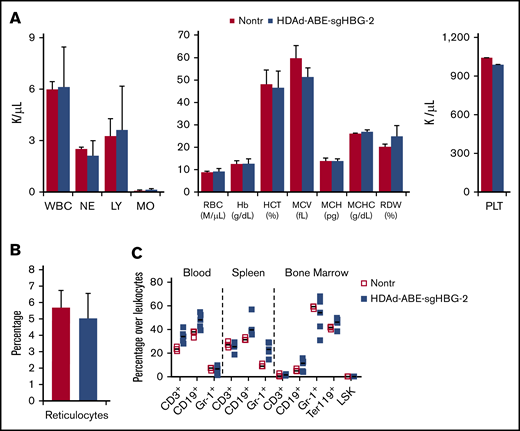
Safety profile. (A) Hematology analysis by Hemavet, using blood samples collected 16 weeks after transduction. Data are means with SD representing 9 mice transduced with HDAd-ABE-sgHBG-2 and 3 nontransduced naive mice (Nontr). (B) Percentage of reticulocytes in blood samples at week 16. The blood smears were stained with Brilliant cresyl blue. (C) Cellular composition of bone marrow mononuclear cells at the terminal point of primary mice. Nontransduced mice were used as the control. Each symbol represents 1 animal. The differences between the groups were not significant.
Safety profile. (A) Hematology analysis by Hemavet, using blood samples collected 16 weeks after transduction. Data are means with SD representing 9 mice transduced with HDAd-ABE-sgHBG-2 and 3 nontransduced naive mice (Nontr). (B) Percentage of reticulocytes in blood samples at week 16. The blood smears were stained with Brilliant cresyl blue. (C) Cellular composition of bone marrow mononuclear cells at the terminal point of primary mice. Nontransduced mice were used as the control. Each symbol represents 1 animal. The differences between the groups were not significant.
To demonstrate that in vivo transduction occurred in long-term repopulating HSPCs, we transplanted bone marrow lineage-negative (Lin–) cells harvested at week 16 after in vivo transduction into lethally irradiated C57BL/6J mice (not expressing the human CD46). The ability of transplanted cells to drive multilineage reconstitution in secondary recipients was evaluated over 16 weeks. Engraftment rates based on huCD46 expression in PBMCs were >95% and remained stable (supplemental Figure 7A). GFP marking of PMBCs was comparable to that in primary mice (supplemental Figure 7B). The average percentage of γ-globin+ RBCs was 40% and stable over time (supplemental Figure 7C). These observations demonstrate that in vivo HSPC base editing was safe overall. The modified HSPCs persisted for the long term and were capable of reconstituting secondary recipient mice with stable transgene expression.
Minimal HBG1/2 intergenic deletion and no detectable editing at top-scoring, off-target sites
A potential problem with DSB-depending gene editing strategies is large genomic deletions and rearrangements that can involve off-target sites.2 This side effect becomes more pronounced when targeting the HBG1/2 promoters. Because of the 2 identical targets in the HBG1 and HBG2 promoter regions, we and others have reported that CRISPR/Cas9 editing led to a deletion of the 4.9-kb region between the 2 sites.17,27 As a result, the whole HBG2 gene was removed. Therefore, we interrogated this genomic deletion by semiquantitative PCR, as described previously.27 For comparison, we included mouse samples treated side by side with a CRISPR/Cas9 vector (HDAd-HBG-CRISPR) targeting the HBG1/2 promoters.27 We found that the average frequency of the 4.9-kb deletion in HDAd-BE-sgHBG-2–treated mice was 0.8% (Figure 6A-B). In some mice, it was barely detectable, an average of 10-fold less than the observed deletions in HDAd-HBG-CRISPR–treated samples.
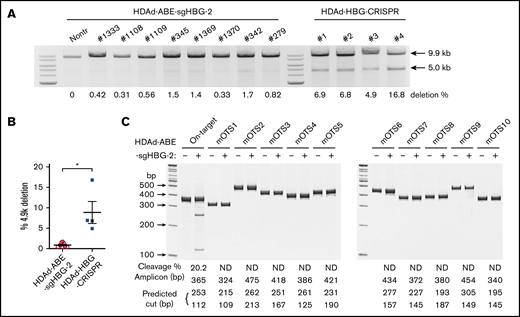
Detection of intergenic deletion. (A) The detection of the intergenic 4.9-k deletion was performed as described previously.27,38 Genomic DNA isolated from total bone marrow cells was used as a template. A 9.9-kb genomic region spanning the 2 sgHBG-2 mapping sites within the HBG1 and HBG2 promoters was amplified by PCR. An additional 5.0-kb band in the product indicates the occurrence of the 4.9-k deletion. The percentage of deletion was calculated according to a standard curve formula that was generated by PCR of templates with defined ratios of the 4.9-k deletion. Samples derived from mice in vivo transduced with an HDAd-HBG-CRISPR vector27 targeting the HBG1/2 promoter were used for comparison. Each lane represents 1 animal. (B) Summary of the percentage of deletion of 4.9 k in panel A. Each symbol represents 1 animal. *P < .05. (C) Measurement of off-target editing by T7EI cleavage assay. The 10 top-scoring, off-target sites in the whole-mouse genome (mOTS-1 to -10; supplemental Tables 1 and 3) were amplified from genomic DNA of bone marrow cells. Samples from nontransduced mice were used as the negative control. The amplicon size and predicted bands after cleavage are listed below the gel lanes. On-target cleavage at the HBG1/2 promoter is also included. The percentage of cleavage is also shown below the lanes. ND, not detected. DNA ladder sizes are indicated on the left of the figure.
Detection of intergenic deletion. (A) The detection of the intergenic 4.9-k deletion was performed as described previously.27,38 Genomic DNA isolated from total bone marrow cells was used as a template. A 9.9-kb genomic region spanning the 2 sgHBG-2 mapping sites within the HBG1 and HBG2 promoters was amplified by PCR. An additional 5.0-kb band in the product indicates the occurrence of the 4.9-k deletion. The percentage of deletion was calculated according to a standard curve formula that was generated by PCR of templates with defined ratios of the 4.9-k deletion. Samples derived from mice in vivo transduced with an HDAd-HBG-CRISPR vector27 targeting the HBG1/2 promoter were used for comparison. Each lane represents 1 animal. (B) Summary of the percentage of deletion of 4.9 k in panel A. Each symbol represents 1 animal. *P < .05. (C) Measurement of off-target editing by T7EI cleavage assay. The 10 top-scoring, off-target sites in the whole-mouse genome (mOTS-1 to -10; supplemental Tables 1 and 3) were amplified from genomic DNA of bone marrow cells. Samples from nontransduced mice were used as the negative control. The amplicon size and predicted bands after cleavage are listed below the gel lanes. On-target cleavage at the HBG1/2 promoter is also included. The percentage of cleavage is also shown below the lanes. ND, not detected. DNA ladder sizes are indicated on the left of the figure.
Next, we conducted off-target analyses to examine the fidelity of our system. In silico analysis showed no potential off-target sites with ≤2-bp mismatches in both the human and mouse genomes. There were 10 and 2 potential off-targets with 3-bp mismatches in humans and mice, respectively. The likelihood of off-target editing at these predicted sites is theoretically low, because all the sites bear at least a 1-bp mismatch in the PAM-proximal half of the protospacer. With 4-bp mismatches, 79 and 74 potential targets in humans and mice, respectively, are predicted (supplemental Tables 1 and 2). Because the study was performed in mice, we amplified the 10 top-scoring mouse genomic sites (2 with 3-bp mismatches; 8 with 4-bp mismatches) from the mice with the highest on-target base installation, followed by Sanger sequencing and T7EI assay. None of these sites exhibited detectable editing (Figure 6C; supplemental Table 3).
No significant cytotoxicity in human CD34+ HSPCs after HDAd-BE vector transduction
We have previously observed that human CD34+ HSPCs transduced with HDAd-CRISPR/Cas9 vectors generate much fewer multilineage progenitor colonies in CFU assays and exhibit impaired engraftment in a xenograft mouse model, indicating significant cytotoxicity caused by CRISPR/Cas9 expression.45 To evaluate potential cytotoxicity by BEs, we transduced human CD34+ HSPCs with HDAd-ABE-sgHBG-2 (MOI of 3000 vp per cell). For side-by-side comparison we included an HBG1/2-targeting CRISPR/Cas9 vector (HDAd-HBG-CRISPR) and a GFP-expressing control vector (HDAd-GFP), which did not contain gene-editing machinery. One day after transduction, the cells were subjected to CFU assays, in vitro erythroid differentiation, and engraftment studies. Consistent with previous findings,45 CFU assays showed that HDAd-HBG-CRISPR transduction significantly decreased the number of colonies (P < .05). In contrast, HDAd-BE-sgHBG-2 transduction showed no effect on colony numbers when compared with HDAd-GFP treatment (Figure 7A). We measured the editing frequency by T7EI assay at various time points after transduction and differentiation. Although the BE vector generated lower editing rates than the CRISPR vector (7% vs 21% at day 4 after transduction), the editing levels remained stable in BE vector-transduced cells, in contrast to the significant decrease in editing after HDAd-HBG-CRISPR treatment (Figure 7B). Engraftment studies were conducted by transplanting transduced CD34+ cells into sublethally irradiated NOD-scid IL2rγnull mice. Results showed that HDAd-BE-sgHBG-2 transduction led to similar levels of engraftment (measured by huCD45 expression) when compared with the HDAd-GFP treatment (Figure 7C), whereas HDAd-HBG-CRISPR transduction largely deprived CD34+ cells of their engraftment capability, in line with previous findings.45 Importantly, the editing level was maintained in engrafted cells harvested at week 8 after transplantation (Figure 7B). Taken together, these data suggest no evident cytotoxicity from HDAd-ABE-sgHBG-2 transduction in human HSPCs.
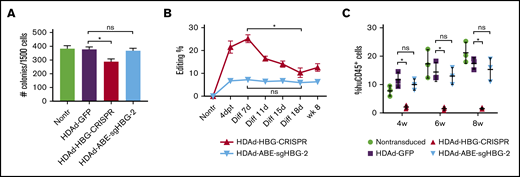
Cytotoxicity of HDAds expressing BEs vs CRISPR/Cas9. Human CD34+ HSPCs were transduced with the CRISPR vector HDAd-HBG-CRISPR, the BE vector HDAd-ABE-sgHBG-2, or a control vector HDAd-GFP at an MOI of 3000 vp per cell. Twenty-four hours after transduction, the cells were used for CFU assay, in vitro erythroid differentiation, and in vivo engraftment studies. (A) Colonies with more than 100 cells (including CFU-E, CFU-G, CFU-GM, BFU-E, CFU-M, and CFU-GEMM) were scored 14 days after plating the transduced cells in methylcellulose supplemented with cytokines and growth factors. The ratio of each colony type was similar among different treatments. Experiments were performed in triplicate. Representative data (mean with SD) of 3 different donors are shown. (B) Target site editing was measured by a T7EI assay at 4 days after transduction (4dpt), various time points after differentiation (Diff), and 8 weeks after transplantation. (C) Cells were IV infused into irradiated NOD-scid IL2rγnull mice at 5 × 105 cells per mouse (n = 3 per treatment). Nontransduced cells or HSPCs transduced with a GFP-expressing vector (HDAd-GFP) were used as controls. Engraftment reflected by percentage of human CD45+ cells in PBMCs at the indicated weeks after infusion was measured by flow cytometry. Each dot represents 1 animal. *P < .05. ns, not significant.
Cytotoxicity of HDAds expressing BEs vs CRISPR/Cas9. Human CD34+ HSPCs were transduced with the CRISPR vector HDAd-HBG-CRISPR, the BE vector HDAd-ABE-sgHBG-2, or a control vector HDAd-GFP at an MOI of 3000 vp per cell. Twenty-four hours after transduction, the cells were used for CFU assay, in vitro erythroid differentiation, and in vivo engraftment studies. (A) Colonies with more than 100 cells (including CFU-E, CFU-G, CFU-GM, BFU-E, CFU-M, and CFU-GEMM) were scored 14 days after plating the transduced cells in methylcellulose supplemented with cytokines and growth factors. The ratio of each colony type was similar among different treatments. Experiments were performed in triplicate. Representative data (mean with SD) of 3 different donors are shown. (B) Target site editing was measured by a T7EI assay at 4 days after transduction (4dpt), various time points after differentiation (Diff), and 8 weeks after transplantation. (C) Cells were IV infused into irradiated NOD-scid IL2rγnull mice at 5 × 105 cells per mouse (n = 3 per treatment). Nontransduced cells or HSPCs transduced with a GFP-expressing vector (HDAd-GFP) were used as controls. Engraftment reflected by percentage of human CD45+ cells in PBMCs at the indicated weeks after infusion was measured by flow cytometry. Each dot represents 1 animal. *P < .05. ns, not significant.
Discussion
In the current study, we demonstrate, for the first time, direct in vivo genome editing with BEs to reactivate fetal hemoglobin. Specifically, we show the generation of the –113A>G HPFH mutation at a conversion rate of 20% in HSPCs of β-YAC mice, leading to >40% γ-globin+ peripheral RBCs. This marking rate was maintained in secondary recipients. Importantly, compared with CRISPR/Cas9, base editing in HSPCs appeared to be safer in both transgenic mice and human CD34+ cells.
We generated vectorized BEs that can be used for in vivo HSPC transduction. We used HDAd5/35++ vectors with chimeric serotype 5/35++ fibers targeting CD46, a receptor that is uniformly expressed on human HSPCs. In addition to efficient transduction of HSPCs, HDAd5/35++ vectors have several advantages, including the >32-kb payload capacity, relatively low cost for large-scale vector production and controllable innate toxicity.37 We used a novel vector design by placing the BE expression cassette outside the SB100× transposon. This design allows for (1) transient expression of the BE, while concurrently maintaining integrated expression of the mgmt/GFP cassette for selection, and (2) more rapid degradation of genes encoding the BE upon coinfection with HDAd-SB.29 Although we were initially unable to produce ABE HDAd vectors, a second version with modified promoters, use of alternative codons and a mi-RNA–regulated gene expression mechanism led to successful generation of ABE vectors. These strategies can be used for generating other HDAd vectors with repetitive sequences and/or requiring transgene suppression during vector production.
Inspired by the protective effect of HbF expression in patients with β-hemoglobinopathy with HPFH mutations, the creation of these mutations by genome editing to reactivate fetal hemoglobin is being pursued as a therapeutic strategy for β-globin disorders. We and several other groups have used endonuclease-based approaches to edit the HBG1/2 promoters and have achieved therapeutically relevant HbF production in transgenic mice and human CD34+ HSPCs.27,42,46-48 As emphasized by Weiss et al, despite its promising efficacy, the editing nature of endonucleases relying on DSBs may cause critical adverse effects, such as p53-dependent DNA damage responses and unwanted large genomic deletions.1-3 Specifically, the simultaneous cleavage at the target sites in both HBG1 and HBG2 promoters leads to a deletion of the 4.9-kb intergenic region containing the whole HBG2 gene.27,42,46,47 Because base editing is not dependent on DSBs, the risk of large deletions may be much lower. Indeed, in total bone marrow cells from mice treated with HDAd-ABE-sgHBG-2 or HDAd-HBG-CRISPR, a magnitude lower frequency (0.8% vs 8%) of the 4.9-kb deletion was found in the animal group treated with the BE-expressing HDAd. It is likely that an even lower frequency of HBG2 deletions, generated by the BE vector, could be achieved by replacing the nCas9 with a catalytically dead Cas9, which is incapable of nicking the nonedited DNA strand. However, this may come at a cost of base editing efficacy.5 Advantages of HDAd-BEs outweigh their relatively lower efficacy in huCD34+ cells compared with CRISPR/Cas9 vectors. The lower frequency could be mitigated by in vivo selection, more active BE variants, or future vectors with combinatorial strategies (for example, base editing+gene addition).
Our study shows that after in vivo HSPC transduction/selection, 20% of total bone marrow cells contained the –113 A>G HPFH mutation. The level of γ-globin expression directly correlated with the percentage of base conversions. The ∼21% chain level of γ-globin over that of β-globin demonstrates potential therapeutic relevance for patients with SCD and β-thalassemia. Notably, because of the disease background, the survival advantage of gene-corrected erythroid progenitors49 could further amplify the therapeutic effect of base editing. It is noteworthy that the current BE constructs contain a GFP reporter that is bicistronically linked with the mgmtP140K gene. Future constructs with a monocistronic design (removal of the GFP gene) could contribute to more efficient mgmtP140K expression. As a result, the editing efficacy and large variation among the animals observed in this study could be further improved.
Other than targeting a single locus, multiplexed editing at both the HBG1/2 promoters and the BCL11A enhancer loci may lead to additive or even synergistic effects on γ-globin expression, as observed in our studies with the dual vector (Figure 2). Furthermore, recent studies have introduced more robust BEs, such as ABE8e50 and ABE8s,51 which should be included in future studies. Although current BEs cannot directly reverse the sickle mutation (GTG>GAG), BEs can generate functional HBB variants, such as the naturally occurring G-Makassar variant (GTG>GCG),52 which would lead to an amelioration of the SCD phenotype. Notably, In addition to BEs, prime editors can generate any types of genomic microalterations,53 including the precise reversal of the sickle mutation; however, their efficacy in HSPCs has yet to be confirmed.
In vivo HSPC base editing was safe in β-YAC mice. We did not observe any significant changes in hematological parameter or cellular composition in peripheral and bone marrow samples. In contrast to Ad5 vectors, the HDAd5/35++ vectors do not transduce hepatocytes after IV delivery, corroborated in both CD46tg mice and nonhuman primates.22,54 Moreover, at the top-10–scoring loci in the mouse genome, no detectable off-target editing was found by our method. Genome-wide off-target effects could be further profiled by using more powerful approaches such as GUIDE-seq.55 Future studies should also examine potential off-target RNA editing, as previously reported.56,57
Compared with conventional ex vivo approaches, the in vivo HSPC transduction approach with HDAd-BE vectors presents advantages in simplicity, safety, and cost-effectiveness. Mobilized HSPCs are transduced with HDAd vectors in the periphery and home back to the bone marrow shortly after transduction.22 The procedure involves no stem cell harvest, no ex vivo manipulation of HSPCs, and no myeloablation. Because of the fiber modification made to HDAd5/35++ vectors and innate immune response prophylaxis, the delivery of vectors by IV injection is well tolerated. In contrast to ex vivo approaches that require intensive procedures and are expensive and therefore difficult to deliver to most patients, the simplified and efficient in vivo strategy may be implemented as an outpatient procedure. Given that most patients with β-globin disorders live in resource-limited regions, in vivo HSPC base editing holds the potential for more affordable and broad applications.
In summary, we present the first study, to our knowledge, of in vivo base editing for γ-globin reactivation in transgenic β-YAC mice. The effectiveness and safety profile of our approach demonstrates potential therapeutic relevance for the treatment of β-hemoglobinopathies.
For original data, please send requests to Chang Li at cli1239@uw.edu.
Acknowledgments
The authors thank Hans-Peter Kiem for helpful comments.
The study was supported by National Institutes of Health, National Heart, Lung, and Blood Institute (grants R01HL128288 and R01HL141781) (A.L.). A.G. was supported by a scholarship from the Foundation of the Hellenic Society of Hematology.
Authorship
Contribution: C.L. and A.L. conceived the study and designed the experiments; C.L., A.G., A.M., and S.G. performed the experiments; C.L. wrote the manuscript; E.Y. and R.D.H. provided critical comments; and A.L. supervised the study.
Conflict-of-interest disclosure: A.L. is a cofounder of Ensoma, Inc. The remaining authors declare no competing financial interests.
Correspondence: Chang Li, University of Washington, PO Box 357720, Seattle, WA 98195; e-mail: cli1239@uw.edu.

