

Key Points
Mutations of residues responsible for extended knob-hole interactions in fibrin result in altered clot formation, structure, and mechanics.
Molecular dynamic simulations show that only slip bonds occur in mutant systems, whereas catch-slip bonds are present in wild-type fibrin.

Abstract
Fibrin polymerization involves thrombin-mediated exposure of knobs on one monomer that bind to holes available on another, leading to the formation of fibers. In silico evidence has suggested that the classical A:a knob-hole interaction is enhanced by surrounding residues not directly involved in the binding pocket of hole a, via noncovalent interactions with knob A. We assessed the importance of extended knob-hole interactions by performing biochemical, biophysical, and in silico modeling studies on recombinant human fibrinogen variants with mutations at residues responsible for the extended interactions. Three single fibrinogen variants, γD297N, γE323Q, and γK356Q, and a triple variant γDEK (γD297N/γE323Q/γK356Q) were produced in a CHO (Chinese Hamster Ovary) cell expression system. Longitudinal protofibril growth probed by atomic force microscopy was disrupted for γD297N and enhanced for the γK356Q mutation. Initial polymerization rates were reduced for all variants in turbidimetric studies. Laser scanning confocal microscopy showed that γDEK and γE323Q produced denser clots, whereas γD297N and γK356Q were similar to wild type. Scanning electron microscopy and light scattering studies showed that fiber thickness and protofibril packing of the fibers were reduced for all variants. Clot viscoelastic analysis showed that only γDEK was more readily deformable. In silico modeling suggested that most variants displayed only slip-bond dissociation kinetics compared with biphasic catch-slip kinetics characteristics of wild type. These data provide new evidence for the role of extended interactions in supporting the classical knob-hole bonds involving catch-slip behavior in fibrin formation, clot structure, and clot mechanics.
Introduction
Fibrin polymerization is a key process in hemostasis that provides a protein scaffold for the blood clot to stem bleeding. Fibrin(ogen) also plays a role in atherothrombosis,1 venous thromboembolism,2 angiogenesis,3 and infection control.4,5 Fibrinogen is comprised of 3 pairs of polypeptides (Aα2Bβ2γ2), linked by disulphide bonds forming a trinodular elongated protein.6,7 The N termini of all chains begin in the central E-region and extend outward in a coiled coil to two distal D-regions. The Bβ- and γ-chains terminate in the D-region, whereas the longer flexible Aα-chain folds backward along the coiled coil toward the E-region.8
The conversion of fibrinogen to fibrin is facilitated by thrombin, which cleaves 2 fibrinopeptides, A (FpA) and B (FpB), from the N termini of the Aα- and Bβ-chains, respectively.9 The removal of FpA and FpB exposes knobs A and B on one fibrin monomer that spontaneously bind to permanently exposed holes a (in the γ-chain) and b (Bβ-chain) on another monomer, respectively,10-12 forming half-staggered fibrin oligomers.13 Polymerization continues, forming larger species of oligomers termed protofibrils. On reaching a length of ∼0.5 µm, protofibrils aggregate laterally to form fibers.14 The current view of fibrin polymerization dynamics is that A:a knob-hole interactions drive longitudinal growth, which results in formation of oligomers and protofibrils, whereas the B:b knob-hole and α-chain connector interactions are responsible for lateral aggregation.15-24
Evidence suggests that A:a interactions involve additional residues beyond the traditional binding pocket (hole a). The synthetic knob A peptide GPRP (Gly-Pro-Arg-Pro), used to recapitulate knob A, weakly binds with hole a in cross-linked double-D fragment (Kd of 25 µM),12 whereas larger fibrin fragments bind ∼5 times more tightly with fibrinogen (Kd of 5.8 µM).25 This provides evidence that additional residues beyond the classical A:a interaction sites contribute to knob-hole coupling. Recent all-atom molecular dynamic (MD) simulation studies provide further evidence that extended interactions occur near the A:a interface,26 indicating that A:a interactions are enhanced by electrostatic interactions between γGlu323/βLys58, γLys356/βAsp61, and γAsp297/βHis67. Furthermore, a recent study combining single-molecule forced dissociation by atomic force microscopy (AFM) with MD simulation-based molecular modeling revealed biphasic catch-slip kinetics of A:a knob-hole bond rupture.27 Dynamic pulling force-induced remodeling of the A:a association interface provided a molecular mechanism underlying such dual-catch-slip response to external mechanical factors.
This body of evidence from experimental and computational modeling studies indicate the existence of additional contacts between residues outside the immediate A:a interface, which might also become activated mechanically. However, no studies have hitherto explored the effects of mitigation of catch-slip-type dynamics in knob-hole interactions on the kinetics of fibrin polymerization and resulting clot structure. To better understand the role of these additional interactions, we produced 4 recombinant human fibrinogens with mutations in the extended interaction sites, γD297N, γE323Q, and γK356Q, and a γDEK variant combining all 3 mutations. We investigated fibrin polymerization and clot structure in conjunction with MD simulations of atomic structural models of the A:a knob-hole complexes. Our studies reveal that γD297 and γE323 are important elements in the transition of the A:a knob-hole interactions from catch to slip bonds, whereas γK356 is primarily involved in the lateral packing interactions at the protofibril level. All variants altered fiber packing and structure, whereas the γDEK variant also altered clot density and mechanics. These data demonstrate a key role for extended knob-hole interactions and catch-slip bonds in fibrin formation, clot structure, and clot mechanical properties. It is understood that fibrin biomechanics are important for clot stability in thrombosis,28,29 therefore, these findings have important implications for thromboembolic diseases.
Methods
Recombinant fibrinogen expression
To disrupt electrostatic interactions because of multiple binary contacts between residues preceding knob A and residues in the γ-nodule outside the binding pocket,26 we selected amino-acid substitutions neutralizing side-chain charge: negatively charged Asp to neutral Asn (γD297N); negatively charged Glu to neutral Gln (γE323Q); and positively charged Lys to neutral Glu (γK356Q). The expression and purification of recombinant human fibrinogen has been described previously.4,30 Single-point mutations γD297N, γE323Q, and γK356Q, and triple-point mutation γD297N/γE323Q/γK356Q (γDEK) were created by site-directed mutagenesis of the pMLP (major late promoter) expression vector containing the fibrinogen γ-chain sequence30 (see supplemental Table 1). Recombinant wild-type (WT) protein was also produced. All constructs were confirmed by sequencing and cotransfected with selection plasmid pMSV-His into CHO (Chinese Hamster Ovary) cells already overexpressing fibrinogen Aα and Bβ chains. Clone selection, expression, and purification were performed as described in the supplemental Methods.
Turbidity
Polymerization of fibrin variants (0.5 mg/mL fibrinogen, 5 mM CaCl2, and 0.1 U/mL thrombin [TBS]) (Tris-buffered saline) was analyzed in 384-well plates (Greiner, Stonehouse, UK) using a Powerwave microtiter-plate reader (Bio-Tek, Swindon, UK) as previously described.31 Optical density was measured (λ = 340 nm) every 12 seconds for 2 hours at 37°C. Three outputs were analyzed from the turbidity profiles: lag phase, maximum optical density (MaxOD), and maximum polymerization rate. Additional turbidity measurements were performed using 0.5 BU/mL reptilase (final concentration) to cleave fibrinopeptide A only.
AFM
AFM was used to study early protofibril formation as previously described17,32 (see supplemental Methods). The polymerization components of the reaction were reduced to focus on the early stages of polymerization.
Confocal and electron microscopy
Fibrin clot structure was analyzed by laser scanning confocal microscopy and scanning electron microscopy (SEM) as described4,30 (see supplemental Methods). Fibrinolysis was studied by confocal microscopy (see supplemental Methods). Polymerization conditions for SEM are chosen as they allow for polymerization of clots that are strong enough to withstand mechanical forces applied to them during the EM sample preparation process.
Protofibril packing
Fibrin clots (0.5 mg/mL fibrinogen, 5 mM CaCl2, and 0.1 U/mL thrombin; TBS) were formed in a polystyrene cuvette (Eppendorf, Stevenage, UK) and immediately sealed with parafilm to prevent dehydration. The clots were formed overnight at room temperature. After clot formation, the clots were scanned between 500 < λ < 800 nm in a Λ35 UV-Vis spectrophotometer (Perkin-Elmer, Cambridge, UK). The average fiber diameter, number of protofibrils, and protofibril distribution within a fiber were determined from the wavelength dependent turbidity of fibrin clots as described.32-34
Micro-rheology of fibrin clots
An in-house micro-rheology device (magnetic tweezers) was used to assess the viscoelastic properties of WT and variant clots as previously described32,35 (see supplemental Methods).
Atomic structural models
Atomic structures of the WT fragment D with hole a (residues γCys139-Val411) and hole a with single-point mutations (γD297N, γE323Q, γK356Q, and γDEK) co-complexed with knob A (residues αGly17-Cys36) were extracted from the structural models of fibrin oligomers.6,36 Atomic models with single point mutations were reconstructed in silico by replacing the corresponding amino-acids, that is, γAsp297toAsn (γD297N), γGlu323toGln (γE323Q), and γLys356toGln (γK356Q). The triple mutant γDEK was reconstructed by introducing all three mutations described above. The obtained models of WT and variant A:a knob-hole complexes were energy minimized, using the steepest descent algorithm, and equilibrated at 25°C with the harmonic constraints imposed on the backbone atoms.
Dynamic force experiments in silico
Due to large system size (entire A:a knob-hole bond complex) and long timescale involved (microseconds), we used GPU-accelerated37,38 all-atom MD simulations in implicit water with the solvent accessible surface area (SASA) model of implicit solvation in conjunction with CHARMM19 force field.39 For each model system (γD297N, γE323Q, γK356Q, and γDEK), five independent runs (each of ∼1-µs duration) of stirred MD simulations were performed. To mimic the experimental AFM measurements, in these simulations, we used the following force protocol: (1) pulling force was applied in the direction perpendicular to the A:a association interface to rupture the A:a bond; (2) the Cα-atom of γLys159 in the γ-nodule (hole a) was constrained; and (3) the Cα-atom of αCys36 (knob A) was tagged through the harmonic spring with stiffness k = 100 pN/nm (virtual cantilever). We used the low-friction limit with damping coefficient of 2.0 ps−1. This does not alter the thermodynamics of noncovalent interactions and allows for more efficient sampling of the conformational space. Profiling the average bond lifetime of noncovalent bonds in the constant-force pulling simulations (force-clamp assays) is computationally expensive, and therefore we used the dynamic force-ramp. To mimic the dynamic force-ramp conditions used in single-molecule experiments on protein-protein complexes and to probe the tension-dependent strength of the A:a knob-hole bond, the applied pulling force f = rft was ramped up with the pulling speed of vt = 104 μm/s. This corresponds to a force-loading rate of rf = kvt. = 10−3 N/s.
Results
Recombinant fibrinogens
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of WT and mutant fibrinogens is shown in supplemental Figure 1. The gel demonstrates bands representative of fibrinogen Aα-, Bβ-, and γA-chains at their expected molecular weights. No additional bands were observed, indicating that the samples were homogeneous and did not contain degradation products.
Extended knob-hole variants increase polymerization rate
There were differences in the polymerization profiles between variant and WT fibrinogens (Figure 1A). Lag phase was similar for all variants compared with WT (Figure 1B). However, maximum polymerization rate Vmax was reduced for all variants compared with WT and significantly so for γDEK (−57%, P < .01; Figure 1C). All variants showed reduced maximum OD compared with WT, which was significant for γDEK (−47%, P < .0001) and γE323Q (−25%, P < .001; Figure 1D). Protofibril growth across the variants was determined using AFM (supplemental Figure 2). The average protofibril length for γD297N (−20%, P < .05) and γK356Q (+15%, P < .05) was significantly different from the WT 10 minutes after fibrin polymerization was initiated (Table 1). A similar turbidity profile was observed for WT and recombinant variants when using reptilase for FpA cleavage only (supplemental Figure 3), indicating that observed effects are because of changes in knob-hole binding rather than thrombin affinity.
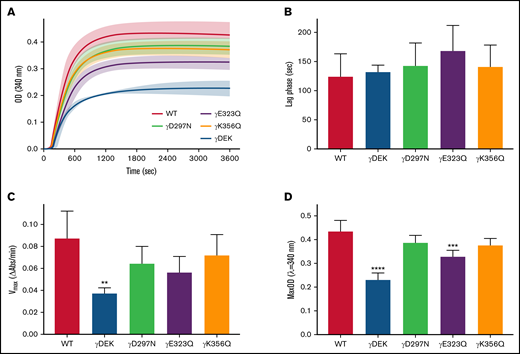
Recombinant fibrinogen variants γDEK, γD297N, γE323Q, and γK356Q corresponding to extended knob-hole interactions affect fibrin polymerization. (A) Polymerization curves, (B) lag phase duration, (C) polymerization rate Vmax, and (D) maximum optical density, MaxOD for all variants and WT fibrinogens. Mean ± standard deviation (SD), N = 3 (γDEK), N = 4 (single variants). **P < .01; ***P < .001, ****P < .0001 compared with WT.
Recombinant fibrinogen variants γDEK, γD297N, γE323Q, and γK356Q corresponding to extended knob-hole interactions affect fibrin polymerization. (A) Polymerization curves, (B) lag phase duration, (C) polymerization rate Vmax, and (D) maximum optical density, MaxOD for all variants and WT fibrinogens. Mean ± standard deviation (SD), N = 3 (γDEK), N = 4 (single variants). **P < .01; ***P < .001, ****P < .0001 compared with WT.
Average protofibril lengths
| 10 min | 20 min | |
|---|---|---|
| WT | 190 ± 71 nm | 250 ± 147 nm |
| γDEK | 163 ± 54 nm | 221 ± 128 nm |
| γD297N | 152 ± 73 nm* | 291 ± 191 nm |
| γE323Q | 184 ± 82 nm | 256 ± 163 nm |
| γK356Q | 218 ± 110 nm* | 286 ± 134 nm |
| 10 min | 20 min | |
|---|---|---|
| WT | 190 ± 71 nm | 250 ± 147 nm |
| γDEK | 163 ± 54 nm | 221 ± 128 nm |
| γD297N | 152 ± 73 nm* | 291 ± 191 nm |
| γE323Q | 184 ± 82 nm | 256 ± 163 nm |
| γK356Q | 218 ± 110 nm* | 286 ± 134 nm |
Results are presented as mean ± standard deviation. Greater than 20 protofibrils were measured per condition over 3 independent experiments.
P < .05 vs WT at the same time point.
Clot structure is influenced by extended knob-hole variants
Next, we analyzed clot density under hydrated conditions using laser scanning confocal microscopy. Clots obtained from γDEK fibrinogen (Figure 2) were significantly denser than those from WT (+39%, P < .001; Figure 2). Visual inspection of the density by scanning electron microscopy of fibrin clot networks at ×20 000 magnification (Figure 3A-E) also displayed a similar trend. All variants resulted in formation of clots with more densely packed fibers characterized by reduced apparent fiber diameter (Figure 3F). No significant differences were observed for fibrinolysis for any of the fibrin clots (supplemental Figure 4).

Density differences of WT and fibrinogen variant fibrin clots involved in extended knob-hole interactions. Fibrin clots were allowed to form for 1 hour and imaged using laser scanning confocal microscopy (A: WT, B: γDEK, C: γD297N, D: γE323Q, E: γK356Q). Fiber count per 200 µm was quantified (F). Variant γDEK (B, F) was denser than WT (A, F). Mean ± SD, N = 4 (WT), N = 3 (all variants). ***P < .001 for variants compared with WT.
Density differences of WT and fibrinogen variant fibrin clots involved in extended knob-hole interactions. Fibrin clots were allowed to form for 1 hour and imaged using laser scanning confocal microscopy (A: WT, B: γDEK, C: γD297N, D: γE323Q, E: γK356Q). Fiber count per 200 µm was quantified (F). Variant γDEK (B, F) was denser than WT (A, F). Mean ± SD, N = 4 (WT), N = 3 (all variants). ***P < .001 for variants compared with WT.

Recombinant fibrinogen variants γDEK, γD297N, γE323Q, and γK356Q affect fiber thickness. Micrographs were taken at ×20 000 magnification using a Hitachi SU8230 scanning electron microscope (A-E). Compared with WT (A), fibrin fibers of γDEK (B, F), γD297N (C, F), γE323Q (D, F), and γK356Q fibers (E, F) were thinner. Mean ± SD, N = 3 (WT, γDEK, γD297N), N = 4 (γK356Q), N = 5 (γE323Q). *P < .05; **P < .01; ***P < .001 compared with WT.
Recombinant fibrinogen variants γDEK, γD297N, γE323Q, and γK356Q affect fiber thickness. Micrographs were taken at ×20 000 magnification using a Hitachi SU8230 scanning electron microscope (A-E). Compared with WT (A), fibrin fibers of γDEK (B, F), γD297N (C, F), γE323Q (D, F), and γK356Q fibers (E, F) were thinner. Mean ± SD, N = 3 (WT, γDEK, γD297N), N = 4 (γK356Q), N = 5 (γE323Q). *P < .05; **P < .01; ***P < .001 compared with WT.
Protofibril packing is increased by extended knob-hole variants
Interestingly, light scattering analysis of fibrin fiber packing demonstrated that extended knob-hole variants led to no apparent change in fiber diameter under fully hydrated conditions, compared with WT (Figure 4A). However, a reduction in average number of protofibrils per fibrin fiber was observed for the recombinant variants (Figure 4B) and was associated with increased distance between individual protofibrils within individual fibers (Figure 4C).
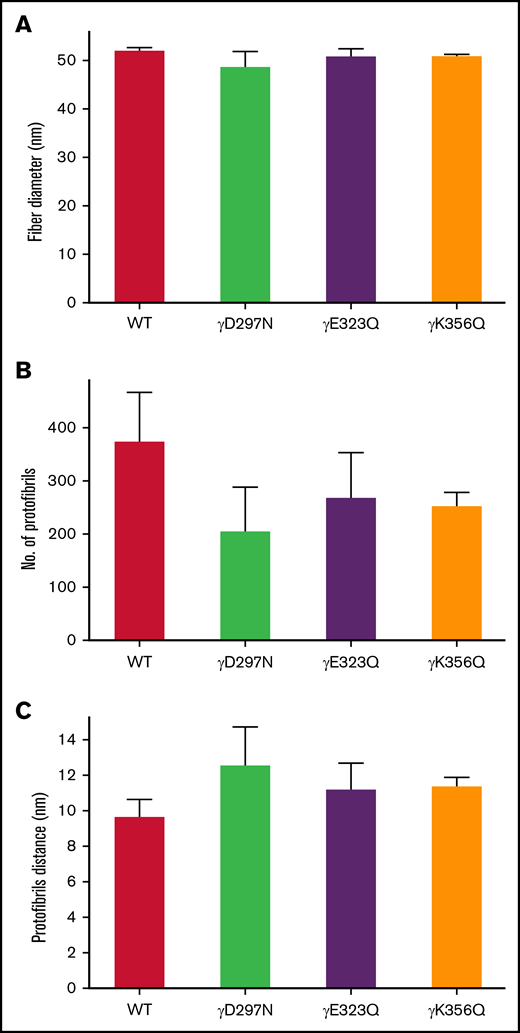
Protofibril packing dynamics of WT and extended knob-hole variant fibrin fibers. The UV-Vis spectrum (500 nm < λ < 780 nm) of fibrin clots was scanned for WT and all variants. The wavelength dependent turbidity of the fibrin clots was used to determine fiber diameter and quantitate intrafibrillar protofibril packing arrangements. The fiber diameter (A) corresponding to all 4 variants were similar to WT, and the number of protofibrils per fiber (B) was reduced for all variants, resulting in an increase in the distance between protofibrils (C). Mean ± SD, N = 3.
Protofibril packing dynamics of WT and extended knob-hole variant fibrin fibers. The UV-Vis spectrum (500 nm < λ < 780 nm) of fibrin clots was scanned for WT and all variants. The wavelength dependent turbidity of the fibrin clots was used to determine fiber diameter and quantitate intrafibrillar protofibril packing arrangements. The fiber diameter (A) corresponding to all 4 variants were similar to WT, and the number of protofibrils per fiber (B) was reduced for all variants, resulting in an increase in the distance between protofibrils (C). Mean ± SD, N = 3.
Extended knob-hole variants support clot mechanics
The viscoelastic properties of fibrin clots were analyzed using magnetic tweezers. The loss tangent (Tan[δ] = G″/G') was calculated at different frequencies to determine the deformation of clots. Three frequencies (0.1, 1, and 10 Hz; Figure 5A-C) were chosen based on deformation events that occur in fibrin networks.32 Tan[δ] at low frequencies represents deformation that occur at the whole clot level, whereas the intermediate-to-high frequency ranges represent deformation at the fibrin fiber level. At 0.1 Hz (Figure 5A), tan[δ] was significantly increased for γDEK (+131%, P < .001) compared with WT, showing that these clots were more readily deformable.
![Extended knob-hole interactions affect overall visco-elastic properties of the clot (tanδ). Micro-rheology experiments were conducted by forming a fibrin clot in a capillary tube. Tan[δ] at 0.1 Hz (A) was significantly increased for γDEK, whereas the single variants were similar to WT. At 1 Hz (B) and 10 Hz (C), [tanδ] was similar to WT for all variants. Mean ± SD, N = 3. ***P < .001 compared with WT.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/13/10.1182_bloodadvances.2022006977/3/m_advancesadv2022006977f5.png?Expires=1767717841&Signature=3kS4dPzfCEZ6jYk4s~fmLudL9iFAb1xetNH6oNqcXl3WY4qZw0H1Fz3Ju5VWIXwm80ch-sLctreoB5kNZIFbgLhtM3UDwNBPp31dwi3iH1jdTJcDFqTh4mWHhF1i1J~M0Qq1wvPXawIojEtPn9c1cVlG13N7FG-jHywLkZYBaghZjYZE1qi~61MtkBDQtl2wxqMaFiyvSwXie1zc9bw3oHWCK4CECctO~NVlRYunkdisXqTFpKpm4mYxb-Y1lM3KRoUIdosQWZ-EXDO~KMmbjG0jtYL0q0ATAMvZu8mhpG7wKK0NzO83ZCUNu6hBsLuRoaQPkiwQd27aLgs9qdvULw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Extended knob-hole interactions affect overall visco-elastic properties of the clot (tanδ). Micro-rheology experiments were conducted by forming a fibrin clot in a capillary tube. Tan[δ] at 0.1 Hz (A) was significantly increased for γDEK, whereas the single variants were similar to WT. At 1 Hz (B) and 10 Hz (C), [tanδ] was similar to WT for all variants. Mean ± SD, N = 3. ***P < .001 compared with WT.
Extended knob-hole interactions affect overall visco-elastic properties of the clot (tanδ). Micro-rheology experiments were conducted by forming a fibrin clot in a capillary tube. Tan[δ] at 0.1 Hz (A) was significantly increased for γDEK, whereas the single variants were similar to WT. At 1 Hz (B) and 10 Hz (C), [tanδ] was similar to WT for all variants. Mean ± SD, N = 3. ***P < .001 compared with WT.
Structural insights from atomic models of fibrin oligomers
Single-point mutations γD297N, γE323Q, and γK356Q are located at the previously identified important binding regions inside and around the binding pocket (hole a), namely in the movable flap (γD297N), loop I (γE323Q), and interior region (γK356Q) (Figure 6). We analyzed the atomic structures of fibrin oligomers and protofibrils reported previously6,36 to estimate the propensities of these residues to form intra- vs inter-protofibril contacts. We found the following: (1) position γ297 in the movable flap is buried in the protofibril, and therefore this residue can mediate formation of the intra-protofibril contacts (promoting the longitudinal growth) but not inter-protofibril contacts; (2) position γ323 in loop I is somewhat exposed to solvent, and hence, this residue can potentially form intra-protofibril contacts and inter-protofibril contacts (promoting lateral packing); and (3) position γK356Q is in the solvent accessible area but far from knob A, and therefore this residue can participate in formation of inter-protofibril contacts.

Computational molecular modeling. (A) Complete atomic structure of the A:a knob-hole complex (in ribbon representation), extracted from the structures of fibrin oligomers6,36 at pH 7.0 and 25°C, showing hole a with the following binding determinants: the interior region (residues γTrp335-Asn365; shown in green), loop I (γTrp315-Trp330; in blue), and movable flap (γPhe295-Thr305; in red) in the γ-nodule, and knob A (residues αGly17-Cys36; shown in orange). Also shown are the setup for the dynamic force experiments in silico, for example, the constrained residue γLys159 in the γ-nodule in hole a and tagged residue αCys36 in knob A, and the direction of pulling force (indicated by the arrow), and single-point mutations corresponding to variants γD297N, γE323Q, and γK356Q (small arrows). (B) The average bond-rupture forces and SDs (mean ± SD, N = 5 simulation runs) for WT and variants γD297N. γE323Q, γK356Q, and γDEK extracted from the simulations. (C-D) The side view (C) and top view (D) of a structural fragment of 2-stranded fibrin oligomer inside its hydrodynamic volume, displaying the α chain (in red color), β chain (in blue), and γ chain (in green). Also shown are the D:D self-association interface, the D:E:D complex with the A:a and B:b knob-hole bonds, the coiled coils, and carbohydrate moieties. These structures reveal the positions of mutated residues relative to the body of fibrin oligomer, which can be correlated with their propensities to form the intra-protofibril vs inter-protofibril contacts. (E) The same structure as in panel D but with another protofibril added to the picture to help visualize the lateral packing with the inter-protofibril contacts corresponding to the crystal contacts in structure 1FZC19 from the protein data bank.
Computational molecular modeling. (A) Complete atomic structure of the A:a knob-hole complex (in ribbon representation), extracted from the structures of fibrin oligomers6,36 at pH 7.0 and 25°C, showing hole a with the following binding determinants: the interior region (residues γTrp335-Asn365; shown in green), loop I (γTrp315-Trp330; in blue), and movable flap (γPhe295-Thr305; in red) in the γ-nodule, and knob A (residues αGly17-Cys36; shown in orange). Also shown are the setup for the dynamic force experiments in silico, for example, the constrained residue γLys159 in the γ-nodule in hole a and tagged residue αCys36 in knob A, and the direction of pulling force (indicated by the arrow), and single-point mutations corresponding to variants γD297N, γE323Q, and γK356Q (small arrows). (B) The average bond-rupture forces and SDs (mean ± SD, N = 5 simulation runs) for WT and variants γD297N. γE323Q, γK356Q, and γDEK extracted from the simulations. (C-D) The side view (C) and top view (D) of a structural fragment of 2-stranded fibrin oligomer inside its hydrodynamic volume, displaying the α chain (in red color), β chain (in blue), and γ chain (in green). Also shown are the D:D self-association interface, the D:E:D complex with the A:a and B:b knob-hole bonds, the coiled coils, and carbohydrate moieties. These structures reveal the positions of mutated residues relative to the body of fibrin oligomer, which can be correlated with their propensities to form the intra-protofibril vs inter-protofibril contacts. (E) The same structure as in panel D but with another protofibril added to the picture to help visualize the lateral packing with the inter-protofibril contacts corresponding to the crystal contacts in structure 1FZC19 from the protein data bank.
Extended interactions strengthen knob-hole bonds
Next, we carried out MD simulations, in which pulling force was applied to knob A to dissociate the A:a bond (Figure 6). The profiles of unbinding force, binding affinity Q, and peak rupture force f were used to investigate the dynamics of bond rupture. The unbinding force quantitates instantaneous mechanical tension in the noncovalent A:a bond. The binding affinity Q reflects the strength of the noncovalent A:a bond. The peak rupture force f corresponds to the tension threshold at which the A:a bond dissociates. The profiles of Q vs f in supplemental Figure 5 show 2 different scenarios: (1) Q first increases and then decreases with force f (catch-slip bond character), and (2) Q decreases monotonically with f (slip bond character). Both catch-slip and slip scenarios were observed for WT and mutants γK356Q, and only slip scenario was observed for mutants γD297N, γE323Q, and γDEK (Figure 7). Therefore, in agreement with insights from the atomic structural models, MD simulations showed the following: (1) the A:a bond weakens and hence dissociates at lower f = 60- to 90-pN rupture force for fibrinogen variants γD297N, γE323Q, and γDEK (in all 5 simulation runs) compared with higher 90- to 120-pN rupture force for WT (from 5 control runs); and (2) the A:a bond weakens (60- to 90-pN rupture force) in 1 of 5 runs and does not change in 4 of 5 runs for fibrinogen γK356Q (supplemental Figure 5). In γD297N, γE323Q, and γDEK, the movable flap did not translocate toward knob A (Figure 7) and therefore did not facilitate additional stabilization to the A:a bond (low-affinity bound state, slip bond character). Hence, mutations in the movable flap (γD297N) and loop I (γE323Q) weaken the A:a bond by about 30 pN. This also explains similar results for triple mutant γDEK (reduced f = 60- to 90-pN rupture force). Mutation γK356Q had less-pronounced effects on the strength of A:a knob-hole bonds, in agreement with structural insights from the atomic models of fibrin oligomers and protofibrils.6,36 The profiles for the average rupture force (Figure 6) summarizes these results, namely the weakening of A:a knob-hole bonds for γD297N, γE323Q, and γDEK variants but near-normal bond for γK356Q.
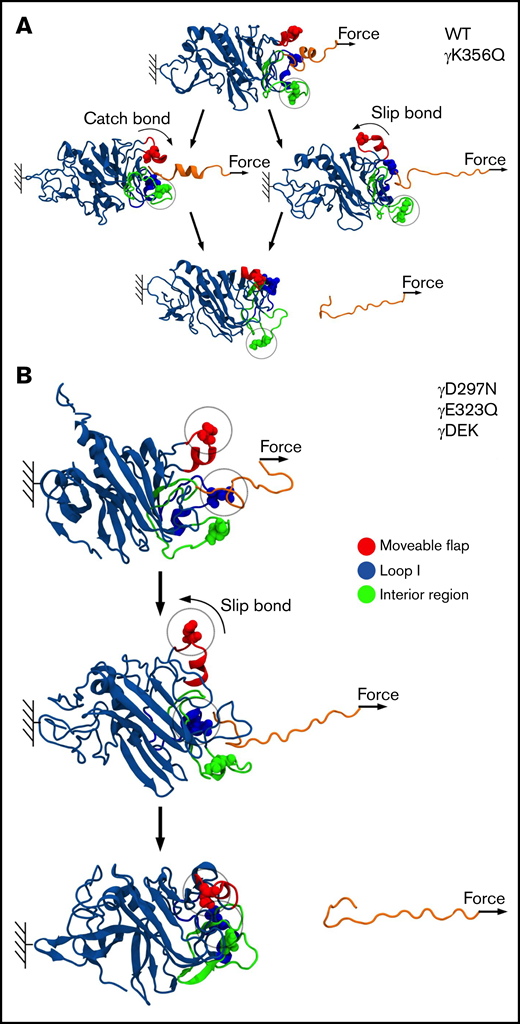
Molecular mechanism of A:a knob-hole bond rupture. Conformational transitions and remodeling of the A:a binding interface are exemplified for single-point mutants γK356Q (green balls) and γD297N (red balls), which result in kinetic partitioning into the bi-phasic catch-slip-type (A) and slip-type bond rupture pathways (B). The atomic structure of the A:a knob-hole complex (in ribbon representation) showing knob A (residues α17-36; in orange color) and hole a with the interior region (γ335-365; in green), loop I (γ315-330; in blue), and movable flap (γ295-305; in red). The pulling force is applied to residue α36 in knob A (small arrows), whereas residue γ159 in the γ-nodule is constrained (color denotation as in Figure 6).
Molecular mechanism of A:a knob-hole bond rupture. Conformational transitions and remodeling of the A:a binding interface are exemplified for single-point mutants γK356Q (green balls) and γD297N (red balls), which result in kinetic partitioning into the bi-phasic catch-slip-type (A) and slip-type bond rupture pathways (B). The atomic structure of the A:a knob-hole complex (in ribbon representation) showing knob A (residues α17-36; in orange color) and hole a with the interior region (γ335-365; in green), loop I (γ315-330; in blue), and movable flap (γ295-305; in red). The pulling force is applied to residue α36 in knob A (small arrows), whereas residue γ159 in the γ-nodule is constrained (color denotation as in Figure 6).
Extended knob-hole interactions enhance formation of catch bonds
A recent study has shown unique and characteristic catch-slip bond behavior for A:a knob-hole interaction.27 Interactions between residues in knob A and residues mainly in the movable flap and, to some extent, in loop I were shown to define dynamic transition from catch to slip bond. In our simulations, the tension-induced strengthening can be detected by analyzing, for example, the pulling force-dependent profiles of the total number of knob-hole residue-residue contacts, which we refer to as binding affinity Q. By multiplying Q by the average contact energy, we obtain the binding energy of the A:a knob-hole interaction.
The force-dependent profiles of Q showed initial increase in Q (high-affinity catch-bond character) in 3 of 5 runs for WT fibrinogen and 1 of 5 runs for mutation γK356Q (supplemental Figure 5). Here, the movable flap extended toward knob A, thus forming additional knob-hole binding contacts between residues γ297-γ304 in the movable flap and residues α17-α22 in knob A (Figure 7). This provides structural support for tension-dependent A:a knob-hole bond stabilization, which results in hole a closure and formation of high-affinity bound state (catch bond). However, for γD297N, γE323Q, and γDEK, Q progressively decreased with force to 0, which corresponds to complete knob-hole bond dissociation, showing the absence of catch-bond formation. Here, negatively charged residues Asp γ291, γ294, γ297, and γ301 in the movable flap (γ291, γ294, and γ301 for γD297N) form strong electrostatic contacts with the positively charged residues Arg γ256 and γ275 in the γ-nodule (supplemental Figure 7). Hence, there is no tension-induced bond stabilization, and only a low-affinity bound state is formed for γD297N, γE323Q, and γDEK (slip bond character).
Discussion
The all-atom MD simulations carried out in this study provide evidence for possible location of extended knob-hole interactions,26 previously postulated by others.40 To test these proposed interaction sites, we produced 4 fibrinogens with mutations in each or all 3 ion-pairing residues that underpin the proposed extended binding. Our study shows that the catch-slip bond behavior of knob A with hole a can be disrupted by variants γD297N, γE323Q, and γDEK that flank the binding pocket. The change in the strength of A:a knob-hole bond and hence the kinetic of binding of knob A to hole a result in reduced fiber growth and decreased protofibril packing, leading to denser clots with reduced fiber mass-length ratio. Changes to residue γK356 involved in lateral inter-protofibril interactions also caused delayed protofibril formation, decreased protofibril packing, and reduced mass-length ratio. The changes from catch-slip to slip character for the A:a bond and disruption of packing interactions in γDEK caused an increase in overall clot deformability, despite a denser clot network. These data show that extended knob-hole interactions are essential for the formation of catch-slip type A:a bonds and for the assembly of fibrin clots resistant to plastic deformation.
Turbidity analysis showed a decrease in maximum OD for all variants, which is reflective of denser clot networks comprised of fibers with reduced mass-length ratio.14,33,41,42 These changes in clot structure were confirmed by confocal microscopy (clot density) and SEM (fiber diameter). Typically, fibrin clots that are highly branched, with high fiber counts and small pores are considered prothrombotic43 and are associated with high thrombin44 and high fibrinogen concentrations.45 These observations suggest that the reduction in average protofibril number packed per fiber for the variants used in this study enables the presence of free protofibrils that are able to readily form additional fibrin fibers and a denser clot network.
The calculation of fibrin radii using light scattering33,34 does not require sample preparation, compared with SEM which requires fixation and dehydration of samples.32 Our results show that the abolition of extended A:a knob-hole interactions resulted in reduced protofibril packing. Analysis of fibers by turbidimetry showed diameters that were ∼3 times larger than those measured by SEM. Dehydration during SEM sample preparation may impact the final micrographs, as fibers are comprised of ∼20% protein and ∼80% water.46 Because of this high-water content, fibrin fibers behave like a hydrogel.47 For densely packed fibers, less contraction occurs during dehydration, but for less densely packed fibers, this contraction may be larger, leading to thinner fibrin fibers.32 Therefore, abolition of extended knob-hole interactions in the variants lead to reduced protofibril packing and fibers with reduced mass-length ration in SEM and light scattering experiments.
Longitudinal protofibril growth analyzed by AFM was reduced for γD297N and increased for γK356Q after 10 minutes of clotting. The initial delay in protofibril growth for γD297N may result from the absence of A:a catch bonds, which would reduce the stability of forming oligomers and would impact lateral protofibril aggregation. The resulting clots would therefore be structurally different from WT clots. In contrast, γK356Q displayed longer protofibrils after 10 minutes. It was previously suggested that residue γK356 is in a region functionally important for lateral γ-nodule packing interactions.19 Disturbance in lateral packing may allow for the formation of longer rather than thicker protofibrils for γK356Q. It is possible that the protofibril length for polymerizing γDEK fibrin was not different because the enhancement of protofibril length from γK356Q counteracted the reduction in length induced by γD297N.
The fibrin network provides mechanical strength to withstand shear stress and prevent bleeding.48-50 MD simulations have been used to predict the range of forces that thrombi are exposed to within the blood.26 The extensibility of fibrin involves unfolding of the coiled-coil regions and γ-chain C-terminal domain.51,52 The lost tangent, a ratio of loss modulus over storage modulus (G″/G'), was increased for γDEK, indicating that these clots were more viscous and, therefore, more readily deformable overall. This is intriguing, because the altered clot structure for γDEK (denser network and smaller pores) would imply that the clot should be stiffer overall, which has been previously described for these types of clot networks.53,54 However, the change of 2 residues involved in the catch-slip bonding character and 1 residue involved in protofibril packing in γDEK likely leads to increased deformation within the fibrin fibers of the variants and the production of weaker clots, resulting from decreased protofibril packaging and increased protofibril slippage despite the denser overall network structure.
Fibrinogen γD297N resides in a γ294-301-residue stretch called movable flap that is associated with low-affinity calcium binding,55 more commonly known as the γ2 site. Previous modifications of the γ2 site via recombinant fibrinogens γD298A and γD301A caused minor functional abnormalities.56 Here we show that γD297N has an effect on the functional properties of fibrinogen, which we attribute to attenuation of the catch regime of the A:a catch-slip bond. In addition, high affinity calcium binding sites that involve residues γD318, γD320, γG324, and γF32257 are affected in fibrinogen Des Moines (γD320 deletion)58 and Vlissingen (γD319 and γD320 deletion),59 thus resulting in abnormal clots highlighting the importance of residues within this region. Not surprisingly, the results of MD simulations showed that γE323Q fibrinogen results in abolition of the catch-slip bonds. Further clinical evidence for the relevance of this region comes from several variants in residues interacting with γD297, γE323, or γK356. Fibrinogen Miami60 (BβD61G) displays delayed onset and rate of polymerization and BβD61has been shown to form inter-protofibril contacts with γK356.26 BβH67 interacts with γD297, and BβH67L (fibrinogen Sumperk) resulted in hypofibrinogenemia associated with bleeding, delayed polymerization, and abnormal clot properties.61 Residue γE323 interacts with Bβ58, which is abnormal in fibrinogen Christchurch V (BβK58 > frameshift41aa-stop) resulting in a truncated β chain,62 but in this case, the clinical phenotype is likely the result of the truncated protein.
The all-atomic MD simulations carried out in this study showed that γD297N and γE323Q result in a lower unbinding force (weaker slip bond character) compared with WT when knob A was pulled from hole a, indicating the abolition of the high-affinity catch bonds.27 However, γK356Q formed catch-slip bonds at a similar rate as WT but persisted to display altered polymerization and clot structure. To investigate this unexpected result, we reconstructed the protofibril structure in silico. We found that γK356Q was relatively exposed to solvent, and any mutation in this region would likely disturb lateral packing interactions at the inter-protofibril level involving residues γ350-360 and γ370-380 as described.19 In respect to variant γDEK, molecular modeling showed absence of the catch-slip bond character that is expected as they are also absent from γD297N and γE323Q. Across most experiments, γDEK was structurally and functionally most different from WT, suggesting a cumulative effect from the disruption of A:a catch-slip bonds through γD297N and γE323Q and the disruption of lateral packing interactions via γK356Q. This leads to a greater impact on polymerization rate, fiber thickness, and clot density, resulting in clots that are more readily deformable. Hence, the results of structural analysis provide new evidence for the role of movable flap in formation of extended binding with knob A, in dynamic regulation of A:a interactions and transition from the catch bonds to slip bonds in fibrin.
Through state-of-the-art in vitro and in silico methods, our data indicate that A:a knob-hole bonds involved in fibrin polymerization are modified via catch-slip bond mechanisms involving residues γD297 and γE323. We found that small changes within the side chains of these key residues influence catch-slip bonds and lower the rupture force of dissociation of knob A from hole a, which subsequently amplifies into changes in overall clot structure and mechanical properties, with implications for clot stability and thus hemostasis and thrombosis. In pathophysiological circumstances, for example, thrombosis, this could be important as the catch-bond character allows the bonds to counterintuitively strengthen under shear stresses from the flowing blood, possibly preventing embolization. Although not involved in the A:a catch bond formation, residue γK356 was implicated in lateral packing, and when both catch-slip bonds and packing interactions are disturbed, the clot mechanics are affected with clots becoming more readily deformable. Together, these results indicate the involvement of extended interactions near non-covalent A:a catch-slip bonds in clot formation and stability. Future studies should explore how these results impact thrombosis and thromboembolism.
Acknowledgments
The authors thank Martin Fuller, University of Leeds, for assistance in critical point drying of SEM samples.
This study was supported by a British Heart Foundation Studentship for N.L.A. (FS/15/37/31513). N.L.A. was also supported by the British Society of Haemostasis and Thrombosis and the University of Leeds for a J-1 Research exchange collaboration visit to the laboratory of Valeri Barsegov (University of Massachusetts Lowell, MA). The R.A.S.A. laboratory is supported by British Heart Foundation Programme (RG/13/3/30104 and RG/18/11/34036 renewal) and project (PG/16/60/32292) grants. The V.B. laboratory is supported by National Institutes of Health grant R01HL148227 and National Science Foundation grant MCB-2027530.
Authorship
Contribution: N.L.A. performed most of the work, alongside C.D., A.Z., S.R.B., H.R.M., and M.M.D.; M.M.D., C.D., N.L.A., S.D.A.C., V.B., and R.A.S.A. designed the study; N.L.A., C.D., A.Z., V.B., and R.A.S.A. wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert Ariëns, Discovery and Translational Science Department, Leeds Institute of Cardiovascular and Metabolic Medicine, University of Leeds, LS2 9JT, UK; e-mail: r.a.s.ariens@leeds.ac.uk.

