

TO THE EDITOR:
There are at least a dozen hereditary hematopoietic malignancy syndromes.1 Three of these syndromes, driven by ANKRD26, ETV6, or RUNX1 germline mutations, share a phenotype of thrombocytopenia, qualitative platelet defects, and an increased lifetime risk of hematopoietic malignancies.2 RUNX1 mutation carriers also experience increased rates of clonal hematopoiesis. In one analysis, 6 of 9 RUNX1 mutation carriers with thrombocytopenia, but no malignancies, had clonal hematopoiesis prior to age 50.3 A second study detected clonal hematopoiesis in 3 of 4 RUNX1 germline mutation carriers without malignancies.4 Germline ANKRD26 or ETV6 mutations phenocopy RUNX1 germline mutations, but the risk of clonal hematopoiesis in ANKRD26 or ETV6 germline mutation carriers is unknown. Understanding the risk of clonal hematopoiesis in ANKRD26 and ETV6 mutation carriers may better inform the clinical surveillance of these patients before they develop malignancies.
To address this knowledge gap, we performed a cross-sectional study to determine the prevalence of clonal hematopoiesis in 11 patients with germline ANKRD26 or ETV6 mutations and thrombocytopenia but no malignancies.3,4 This is the largest study of clonal hematopoiesis in patients with hereditary thrombocytopenia/hereditary hematopoietic malignancy syndromes to date. The penetrance of malignancies is lower in ANKRD26 (8%) and ETV6 (33%) germline mutation carriers than in RUNX1 mutation carriers (44%), so we hypothesized clonal hematopoiesis would be less prevalent in ANKRD26 or ETV6 mutation carriers relative to RUNX1 mutation carriers.2 All pathogenic/likely pathogenic ANKRD26 variants are located in the 5′ untranslated region of ANKRD26, where the RUNX1 transcription factor binds to suppress expression of ANKRD26 (supplemental Figure 1).5 Therefore, we also hypothesized that ANKRD26 and RUNX1 germline mutation carriers would develop somatic mutations in a similar spectrum of genes.
We enrolled 12 patients from unrelated families (supplemental Figure 2) on institutional review board–approved protocols at the University of Chicago or the Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, Brazil (supplemental Table 1). The study was performed according to the Declaration of Helsinki. Seven unaffected ANKRD26 mutation carriers and 4 unaffected ETV6 mutation carriers enrolled. Patient ages ranged from 8 to 63 years (median, 38 years). We sequenced 1 ANKRD26 mutation carrier with acute myeloid leukemia (AML) to compare clonal hematopoiesis-related and malignancy-related mutations in ANKRD26 mutation carriers (Table 1; supplemental Table 1). Each germline variant was classified using Association for Molecular Pathology and American College of Human Genetics and Genomics criteria.6 Samples included germline (cultured skin fibroblasts) or hematopoietic tissue equivalents (peripheral blood, bone marrow, or saliva). Panel-based sequencing was performed at the University of Chicago as described previously.7 Details regarding sample processing and sequencing are provided in supplemental Methods.
Germline mutations and somatic driver mutations identified in each individual in ANKRD26- and ETV6-mutated hereditary thrombocytopenia/hereditary hematopoietic malignancy phenocopy cohort
| Germline gene of interest | Variants | Individual | Family | Phenotype | Age(s) at sample collection (years) |
|---|---|---|---|---|---|
| ETV6 | p.R369Q NM_001987.4 | III_3 | Family 1 | Thrombocytopenia | 59, 62, 63 |
| ETV6 | p.R369Q NM_001987.4 | IV_4 | Family 1 | Thrombocytopenia | 36, 38 |
| ETV6 | p.R369Q NM_001987.4 | IV_5 | Family 1 | Thrombocytopenia | 33 |
| ETV6 | p.R369Q NM_001987.4 | III_5 | Family 1 | Thrombocytopenia | 62 |
| ANKRD26 | 5′ UTR c.-118C>T NM_014915.2 | III_2 | Family 2 | Thrombocytopenia | 56, 60 |
| ANKRD26 | 5′ UTR c.-118C>T NM_014915.2 | III_4 | Family 2 | Thrombocytopenia | 55 |
| ANKRD26 | 5′ UTR c.-118C>T NM_014915.2 | IV_1 | Family 2 | Thrombocytopenia | 25 |
| ANKRD26 | 5′ UTR c.-119C>G NM_014915.2 | IV_3 | Family 3 | Thrombocytopenia | 43, 44 |
| ANKRD26 | 5′ UTR c.-119C>G NM_014915.2 | V_1 | Family 3 | Thrombocytopenia | 13 |
| ANKRD26 | 5′ UTR c.-128G>A NM_014915.2 | III_14 | Family 4 | Thrombocytopenia | 22 |
| ANKRD26 | 5′ UTR c.-128G>A NM_014915.2 | II_12 | Family 4 | AML (23% blasts) | 48 |
| ANKRD26 | 5′ UTR c.-128G>A NM_014915.2 | III_16 | Family 4 | Thrombocytopenia | 8 |
| Germline gene of interest | Variants | Individual | Family | Phenotype | Age(s) at sample collection (years) |
|---|---|---|---|---|---|
| ETV6 | p.R369Q NM_001987.4 | III_3 | Family 1 | Thrombocytopenia | 59, 62, 63 |
| ETV6 | p.R369Q NM_001987.4 | IV_4 | Family 1 | Thrombocytopenia | 36, 38 |
| ETV6 | p.R369Q NM_001987.4 | IV_5 | Family 1 | Thrombocytopenia | 33 |
| ETV6 | p.R369Q NM_001987.4 | III_5 | Family 1 | Thrombocytopenia | 62 |
| ANKRD26 | 5′ UTR c.-118C>T NM_014915.2 | III_2 | Family 2 | Thrombocytopenia | 56, 60 |
| ANKRD26 | 5′ UTR c.-118C>T NM_014915.2 | III_4 | Family 2 | Thrombocytopenia | 55 |
| ANKRD26 | 5′ UTR c.-118C>T NM_014915.2 | IV_1 | Family 2 | Thrombocytopenia | 25 |
| ANKRD26 | 5′ UTR c.-119C>G NM_014915.2 | IV_3 | Family 3 | Thrombocytopenia | 43, 44 |
| ANKRD26 | 5′ UTR c.-119C>G NM_014915.2 | V_1 | Family 3 | Thrombocytopenia | 13 |
| ANKRD26 | 5′ UTR c.-128G>A NM_014915.2 | III_14 | Family 4 | Thrombocytopenia | 22 |
| ANKRD26 | 5′ UTR c.-128G>A NM_014915.2 | II_12 | Family 4 | AML (23% blasts) | 48 |
| ANKRD26 | 5′ UTR c.-128G>A NM_014915.2 | III_16 | Family 4 | Thrombocytopenia | 8 |
Family numbers and individual IDs reference pedigrees shown in supplemental Figure 2
Among 7 patients with ANKRD26 mutations, 1 had clonal hematopoiesis driven by a somatic SF3B1 mutation (p.Lys700Glu). This mutation increased from a variant allele frequency of 9.4% at age 56 to 17.4% at age 60 (Table 1; Figure 1). SF3B1 p.Lys700Glu is a known somatic mutation observed in 2.1% of Catalogue of Somatic Mutations in Cancer hematopoietic malignancies.8 The patient with clonal hematopoiesis was also the oldest patient in the ANKRD26 cohort. No ETV6 mutation carriers (n = 4) had detectable clonal hematopoiesis. Unlike RUNX1 mutation carriers, no ANKRD26 or ETV6 mutation carriers under age 50 had detectable clonal hematopoiesis, despite nearly half of samples being collected from individuals in this age group (Figure 1).3,4
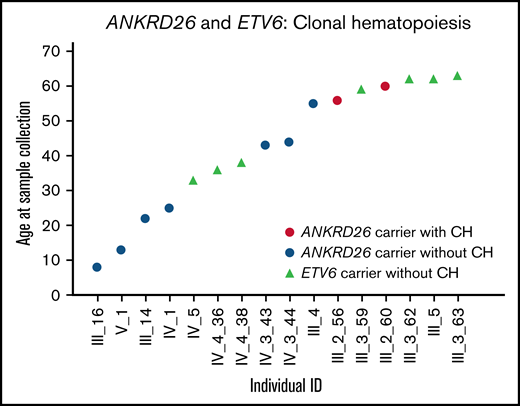
Clonal hematopoiesis in ANKRD26 or ETV6 germline mutation carriers. One individual with a germline ANKRD26 mutation (5′ UTR, c.-118C>T, NM_014915.2) had a CH clone driven by SF3B1 p.Lys700Glu in 2 samples collected at the ages of 56 and 60.
Clonal hematopoiesis in ANKRD26 or ETV6 germline mutation carriers. One individual with a germline ANKRD26 mutation (5′ UTR, c.-118C>T, NM_014915.2) had a CH clone driven by SF3B1 p.Lys700Glu in 2 samples collected at the ages of 56 and 60.
One ANKRD26 mutation carrier had AML, with a karyotype of 46, XX, −6, del(7)(q11.2),+mar[20] and both typical and atypical driver mutations: CUX1 p. Phe472GlnfsX105, RUNX1 p.Arg320X, TET2 p.Phe1309LeufsX54, FLT3 p.Asp835His, and SAMD9 p.Val798GlyfsX7 (Table 1). RUNX1 somatic mutations are the most common second hit in RUNX1 germline mutation carriers with hematopoietic malignancies.8,9 Given that RUNX1 regulates ANKRD26 expression, the RUNX1 mutation in this ANKRD26 mutation carrier may effectively represent a second hit.5
The total observation time for the ANKRD26 cohort was 275 years. The incidence of clonal hematopoiesis in the ANKRD26 cohort was 4.5 clonal hematopoiesis cases per 1000 observation years. The incidence of hematopoietic malignancies in the ANKRD26 cohort was 3.6 malignancies per 1000 observation years. This incidence was similar to an Italian cohort of ANKRD26 mutation carriers (2.1 malignancies per 1000 observation years).10 The observation time for the ETV6 cohort was 258 years, with no clonal hematopoiesis or hematopoietic malignancies.
Among the known hereditary thrombocytopenia/hereditary hematopoietic malignancy phenocopies, only RUNX1-driven syndromes have been evaluated for clonal hematopoiesis risk. This bias has likely occurred for 2 reasons. First, RUNX1 syndromes were identified 12 years before ANKRD26 syndromes and 16 years before ETV6 disorders. This time range provided a longer period for researchers to identify, collect, and study samples from RUNX1 mutation carriers.2,11-13 Second, RUNX1-driven syndromes have the highest cancer penetrance (44%) among the hereditary thrombocytopenia/hereditary hematopoietic malignancy phenocopies.2 In our clinical experience, the most severe hereditary hematopoietic syndromes are more readily recognized than syndromes with subtle symptoms and lower penetrance phenotypes. It is not surprising, therefore, that work in the hereditary hematopoietic malignancy field has focused on RUNX1-driven processes.
In summary, this is the first study of clonal hematopoiesis in the ANKRD26 or ETV6 hereditary thrombocytopenia/hereditary hematopoietic malignancy phenocopies. This study was notable for 3 findings. First, the prevalence of clonal hematopoiesis in the hereditary thrombocytopenia/hereditary hematopoietic malignancy phenocopies is highly variable. Clonal hematopoiesis was detected in 14% of ANKRD26 germline mutation carriers, but no clonal hematopoiesis was detected in ETV6 germline mutation carriers. The overall rate of clonal hematopoiesis in our cohort was 9.1% (95% confidence interval, −8.7% to −27.0%), which is significantly lower than rates in RUNX1 mutation carriers (66% to 75% in Churpek et al and DiFilippo et al, respectively).3,4 Second, early-onset clonal hematopoiesis before age 50 is common in RUNX1 mutation carriers but is uncommon in ANKRD26 or ETV6 mutation carriers.3 Third, the only patient with clonal hematopoiesis in our cohort was observed for over 4 years without changes in their peripheral blood counts. The patient also did not experience leukemogenesis. This finding demonstrates that clonal hematopoiesis in ANKRD26 mutation carriers is, at times, a relatively indolent process that does not portend imminent leukemogenesis.
Our study also had limitations. First, the small number of families in this study may not reflect the risk for clonal hematopoiesis and/or leukemogenesis in larger cohorts of ANKRD26 or ETV6 mutation carriers. However, our cohort of 11 unaffected carriers was larger than prior studies of RUNX1 mutation carriers that were sufficient to detect high rates of clonal hematopoiesis (Churpek et al included 9 unaffected mutation carriers, and DiFilippo et al included 4 unaffected carriers).3,4 Larger numbers of germline ANKRD26 or ETV6 mutation carriers should be analyzed to better determine the preleukemic hematopoietic milieu in these syndromes. Our study was powered (α of 0.05, power of 80%) to detect clonal hematopoiesis at a prevalence as low as 9.1%. However, a second limitation of our work is this cohort is not large enough, given its low prevalence of clonal hematopoiesis, to determine if clonal hematopoiesis in ANKRD26 or ETV6 mutation carriers is significantly higher than similarly aged population controls.
Future studies of acquired mutations before and after the development of hematopoietic malignancies in ANKRD26 or ETV6 germline mutation carriers should be performed to better understand leukemogenic mechanisms in these phenocopies. These studies may ultimately inform the development of the first therapies specifically designed for hereditary thrombocytopenia/hereditary hematopoietic malignancy phenocopies.
Acknowledgments: The authors thank the patients and their families for their participation in this research program and for providing samples.
This work was supported by the Damon Runyon Cancer Research Foundation Physician-Scientist Training Award, the Edward P. Evans Foundation Young Investigator Award, the Cancer Research Foundation Young Investigator Award, and the National Institutes of Health (NIH) Paul Calabresi K12 Program in Oncology (M.W.D.). K.Y., E.K., R.S., and P.P.L. are supported by the Intramural Research Program at NIH National Human Genome Research Institute.
Contribution: M.W. D., C.C.H., K.Y., A.L.B., P.P.L., and L.A.G. designed research; M.W.D., C.C.H., K.Y., K.E.M., M.J.P., M.G.A.-M., J.P.S., P.W., J.G., S.L.K.S., E.K., D.M.L., A.W.S., H.S.S., N.I.S.C., A.L.B., P.P.L., and L.A.G. contributed analytical tools/bioinformatics support; and all authors analyzed data, wrote the paper, and performed research.
Conflict-of-interest disclosure: M.W.D. discloses consulting for Cardinal Health, Inc; H.S.S. discloses honoraria from Celgene; and L.A.G. discloses honoraria from UpToDate. The remaining authors declare no competing financial interests.
Correspondence: Michael W. Drazer, 5841 S. Maryland Avenue, MC 2115, Chicago, IL 60637; e-mail: mdrazer@medicine.bsd.uchicago.edu.

