

Key Points
CAMK2G inhibition ameliorates MF, lessens splenomegaly and leukocytosis, and enhances survival of MF mice induced by MPL W515L.
CAMK2G inhibition can be a novel therapeutic strategy to overcome drug resistance.

Abstract
Although JAK1/2 inhibition is effective in alleviating symptoms of myelofibrosis (MF), it does not result in the eradication of MF clones, which can lead to inhibitor-resistant clones emerging during the treatment. Here, we established induced pluripotent stem cells (iPSCs) derived from MF patient samples (MF-iPSCs) harboring JAK2 V617F, CALR type 1, or CALR type 2 mutations. We demonstrated that these cells faithfully recapitulate the drug sensitivity of the disease. These cells were used for chemical screening, and calcium/calmodulin-dependent protein kinase 2 (CAMK2) was identified as a promising therapeutic target. MF model cells and mice induced by MPL W515L, another type of mutation recurrently detected in MF patients, were used to elucidate the therapeutic potential of CAMK2 inhibition. CAMK2 inhibition was effective against JAK2 inhibitor-sensitive and JAK2 inhibitor-resistant cells. Further research revealed CAMK2 γ subtype was important in MF model cells induced by MPL W515L. We showed that CAMK2G hetero knockout in the primary bone marrow cells expressing MPL W515L decreased colony-forming capacity. CAMK2G inhibition with berbamine, a CAMK2G inhibitor, significantly prolonged survival and reduced disease phenotypes, such as splenomegaly and leukocytosis in a MF mouse model induced by MPL W515L. We investigated the molecular mechanisms underlying the therapeutic effect of CAMK2G inhibition and found that CAMK2G is activated by MPL signaling in MF model cells and is an effector in the MPL-JAK2 signaling pathway in these cells. These results indicate CAMK2G plays an important role in MF, and CAMK2G inhibition may be a novel therapeutic strategy that overcomes resistance to JAK1/2 inhibition.
Introduction
Myelofibrosis (MF) is a myeloproliferative neoplasm characterized by megakaryocytic atypia, fibrosis in the bone marrow (BM), and extramedullary hematopoiesis.1 MF is a preleukemic disease with a poor prognosis, and the median life expectancy of patients ranges from 27 to 135 months.2 Mutational analyses revealed that ∼90% of patients with primary MF have 1 of the 3 mutations, JAK2, CALR, and MPL, which constitutively activate the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. However, these mutations cannot fully explain the molecular pathogenesis of MF.3 Currently, the only effective treatment of MF is allogeneic stem cell transplantation.4 However, allogeneic stem cell transplantation is associated with high treatment-related mortality and morbidity, and most patients are not candidates for transplantation because of their age and comorbidities. Ruxolitinib and fedratinib are JAK1/JAK2 inhibitors currently available for the treatment of MF, and several clinical trials have demonstrated that JAK1/JAK2 inhibitors reduce spleen volume, improve symptoms related to MF, and prolong the overall survival.5-10 Although the benefits associated with JAK1/JAK2 inhibitors are well established, not all patients respond to these inhibitors, and long-term exposure to these inhibitors results in the emergence of resistant clones based on the reactivation of the JAK-STAT pathway.11 Therefore, new therapeutic strategies targeting other molecules can provide additional benefits to patients with MF.
Murine models have been used in several studies on the pathogenesis of MF. However, these models may not fully replicate the human MF. Although theoretically, primary patient samples should be used for research, it is difficult to obtain primary samples from patients with MF owing to pancytopenia caused by fibrosis in the BM. Only a limited number of humanized disease models are currently available to develop novel therapeutic strategies. One promising humanized model would be hematopoietic cells derived from induced pluripotent stem cells (iPSCs) of MF patient samples (MF-iPSCs), as we can expand MF-iPSCs and obtain abundant samples repeatedly after differentiation.12-16 We have previously reported that iPSCs established from MF patients (MF-iPSCs) using retroviral vectors recapitulated the disease phenotype in terms of the expression profile of cytokines.17 To avoid the genomic integration of reprogramming factors and to minimize bias through the reprogramming process, we established integration-free MF-iPSCs from 3 different patients with MF harboring JAK2 V617F, CALR type 1, or CALR type 2 mutations. Using MF-iPSCs, MF model cells overexpressing MPL W515L, and MF mouse model induced by MPL W515L, we comprehensively explored therapeutic targets for MF and demonstrated that the inhibition of calcium/calmodulin-dependent protein kinase 2 (CAMK2) selectively suppressed MF cells both in vitro and in vivo.
Methods
Patient samples
Primary samples from peripheral blood or the BM of all patients were obtained after informed consent. All experiments using human cells were reviewed and approved by the institutional review boards of the University of Tokyo. Mononuclear cells were isolated by Ficoll gradient centrifugation. Characteristics of all patients are described in supplemental Table 1.
Animals
BALB/cCrSlc mice were purchased from Japan SLC, Inc. All mice we used were aged 8 to 12 weeks. All animal experiments were performed in accordance with guidelines for animal experiments of the University of Tokyo.
CAMK2G knockout mice were provided by Eric Olson (University of Texas Southwestern Medical Center, Dallas, Texas) and Johannes Backs (University of Heidelberg, Heidelberg, Germany).18
Cell lines
Ba/F3 cells were purchased from RIKEN BRC, and 32D cells were purchased from JCRB Cell Bank. The mouse C3H10T1/2 cells were cultured as previously descried.13
Flow cytometry
Isolation of each fraction from hematopoietic cells derived from iPSCs, BM cells from mice, and primary samples from patients with MF was performed using FACSAria II and FACSAria III cell sorter (BD Biosciences). Data were analyzed with FlowJo (TreeStar, Ashland, OR). Antibodies are listed in supplemental Table 2.
Generation of iPSCs from MF samples with episomal vectors
To establish iPSCs from MF patient samples, purified CD34+ cells from peripheral blood or the BM of patients with MF were expanded for 2 days in an α-minimum essential media (α-MEM) supplemented with 20% fetal calf serum (FCS), stem cell factor (SCF), ligand for fms-like tyrosine kinase 3 ligand, interleukin (IL)-3, IL-6, and thrombopoietin (TPO), as previously described.16 The plasmid mixture containing pCXLE-hOCT3/4-shp53-F, pCXLE-hSK, pCXLE-hUL, and pCXWB-EBNA1 was electroporated into 2 × 105 CD34+ cells, as described previously.19 The cells were then cultured with mouse embryonic fibroblast for 20 to 30 days, and we obtained embryonic stem (ES) cell-like colonies from 3 different patients.
RNA extraction, real-time quantitative PCR, 1-step RT-PCR analysis
After extraction of total RNA with RNAeasy reagents (QIAGEN), reverse transcription was performed with ReverTra Ace quantitative polymerase chain reaction (qPCR) RT Master Mix (TOYOBO). PCRs were carried out in the LightCycler480 system (Roche) with THUNDERBIRD SYBR qPCR Mix according to the manufacturer’s instructions (TOYOBO). Each assay was performed in triplicate, and the results were normalized to 18S ribosomal RNA. Primer sequences are listed in supplemental Table 3.
Characterization of MF-iPSCs clones
Semiquantitative reverse transcription PCR (RT-PCR) for stem cell genes was performed with the primers described in the literature.20 Immunofluorescence staining was performed for expressions of stage-specific embryonic antigen-4 (SSEA-4) and tumor-related antigen-1-60 (TRA-1-60) with immunofluorescence microscopy as previously described.16 Teratoma formation assay was performed by the injection of 1.0 × 106 cells in the testis capsule of NSC mice. Ten weeks after injection, tumors were harvested and fixed with 3.8% formaldehyde. The fixed tumors embedded in paraffin were stained with hematoxylin and eosin for histological analyses. Karyotype was determined by the G-banding method (Nihon Gene Research Laboratories).
Hematopoietic differentiation from iPSCs
For hematopoietic differentiation from iPSCs, we adopted the protocols described in the literature.21 In brief, clusters of iPSCs were transferred onto C3H10T1/2 cells treated by mitomycin C and cocultured for hematopoietic differentiation in the Iscove’s modified Dulbecco media containing a cocktail of 10 mg/mL human insulin, 5.5 mg/mL human transferrin, 5 ng/mL sodium selenite, 2 mmol/L l-glutamine, 0.45 mmol/L monothioglycerol, 50 mg/mL ascorbic acid, and 15% highly filtered fetal bovine serum in the presence of 20 ng/mL human vascular endothelial growth factor. A cell culture medium was replaced on days 4, 7, 10, 12, and 14. After 14 to 15 days of culture, the iPSC sacs crashed with pipette tips were filtered with cell strainer, and CD34+/CD43+ hematopoietic progenitor cells (HPCs) were isolated by fluorescence-activated cell sorting, as described above.
Compound screening
Screening Committee of Anticancer Drugs inhibitor kit 1 (version 3.1) and kit 2 (version 1.4) were provided from Screening Committee of Anticancer Drugs. All compounds were added at the concentration of 0.1, 1, and 10 μM. The compounds that inhibited MF significantly more than normal cells at all concentrations were given a score of 2. The compounds that inhibited MF cells more than normal cells at 2 sequential concentrations (0.1 μM and 1 μM, or 1 μM and 10 μM) were given a score of 1. The others were given a score of 0.
ATP-luciferase viability assay
Three hundred purified HPCs derived from iPSCs were suspended in α-MEM containing 20% FCS supplemented with 100 ng/mL SCF, 10 ng/mL recombinant megakaryocyte growth and development factor, and 10 ng/mL IL-3. The suspension was placed into a 384-well plate (Greiner Bio-One) and was incubated at 37°C in a 5% CO2 incubator for 48 hours. Subsequently, ATP-luciferase (TOYO INK) was added in each well following manufacturer’s protocol, and the luminescence was measured as relative light unit (RLU) by TriSter2 LB942 (BEERTHOLD). The cell viability was calculated by the formula as follows: cell viability (%) = 100 × (RLU from each well − RLU from background)/(RLU from control [dimethyl sulfoxide (DMSO)] − RLU from background).
Proliferation assay
In each experiment, Ba/F3 and 32D cells were cultured in the RPMI 1640 containing 10% FCS supplemented with or without a 10% WEHI-3B supernatant. Each assay was performed in duplicate.
Apoptosis assay
Apoptosis assay was performed in the same media as proliferation assay in the Ba/F3 cell line. Forty-eight hours after exposure to compounds, cells were stained with annexin V APC and 4,6-diamidino-2-phenylindole following manufacturer’s protocol. In mice peripheral blood (PB), cells were stained with annexin V APC and propidium iodide following manufacturer’s protocol.
Colony-forming cell assay
In each experiment, five hundred CD34+ cells from peripheral blood of MF patients or five hundred purified c-kit+ murine cells transduced with vectors were plated onto a MethoCult GF M4434 and M3434 medium (Stemcell Technologies), respectively. The number of colonies in each experiment was scored on day 7 following manufacturer’s instructions.
Plasmids
The plasmid containing the sequence of FLAG-tagged human CAMK2G was purchased from Invitrogen. To produce protein-expressing retrovirus, we used the following plasmids: pGCDNsam-IRES-GFP or pGCDNsam-IRES-Kusabira-Orange, pMSCV puro. All constructed plasmids were verified by DNA sequencing.
Retrovirus production and transduction of cells
To obtain retroviral supernatants, Plat-E packaging cells were transiently transfected with pGCDNsam vectors, pMSCV puro or pSIREN-RetroQ-ZsGreen containing short hairpin RNA (shRNA) sequences (Addgene). Forty-eight hours later, the viral supernatant was collected and used for infection. The vector-transduced cells were purified by fluorescence-activated cell sorting (FACS) and subjected to assays in vitro. The target sequences, from 5' to 3', were TGCCGACTTCTGAAACATC (CAMK2G sh_1) and TGGAAAACCTGTGGACATC (CAMK2G sh_3), CGTGGAATTTATGCGAATGAT (shJAK_1), CCAACATTACAGAGGCATAAT (shJAK_2).
Immunoblotting
Cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting. Membranes were probed with the following antibodies: anti-FLAG (Sigma), anti-CAMK2G (Cell Signaling Technology), anti-phospho-CAMK2G (Cell Signaling Technology), anti-JAK2 (Cell Signaling Technology), anti-phospho-JAK2 (Cell Signaling Technology), anti-STAT5, anti-phospho-STAT5, and anti-β-actin (Cell Signaling Technology). Blots were detected using an ImmunoStar ζ (Wako Pure Chemical Industries) and LAS-4000 image analyzer (Fujifilm), as recommended by the manufacturers.
Replating assay
In each experiment, three hundred purified c-kit+ murine cells transduced with vectors were plated onto a MethoCult M3434 medium (STEMCELL Technologies). After 7 days of culture, ten thousand cells were replated onto a fresh M3434 medium and proceeded to the next round. The number of colonies in each round was scored on day 7 following manufacturer’s instructions.
Retrovirus production and a murine model of MPL
To obtain retroviral supernatants, Plat-E packaging cells were transiently transfected with pGCDNsam-huMPL wt-IRES-GFP and pGCDNsam-huMPL W515L-IRES-GFP. C-Kit+ BM cells of BALB/cCrSlc mice were purified and collected by autoMACS (Myltenyi Biotec) and incubated in α-MEM with 20% FCS, 1% PS, and cytokines (50 ng/mL SCF, 50 ng/mL TPO, 10 ng/mL IL-6) at 37°C in a 5% CO2 incubator for 24 hours, as previously described.22 Subsequently, cultured cells were transduced with retroviruses in the presence of RetroNectin (Takara Bio Inc). The infected cells were collected 48 hours after retroviral infection, and vector-transduced cells were injected (2.5 × 105 cells per animal) into lethally irradiated (9.5 Gy) recipient mice.
Immunofluorescence analysis
A total of 2 × 104 to 5 × 104 Ba/F3 and 32D cells expressing wild-type MPL or MPL W515L were cytospun onto glass slides. The cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 30 minutes, permeabilized by 0.2% Triton X in PBS for 10 minutes, and blocked with 1% bovine serum albumin in PBS for 60 minutes. Then, the slides were incubated with rabbit anti-CAMK2G polyclonal antibody (ab37999, 1:100 dilution; Abcam) overnight at 4°C, followed by incubation with Alexa Fluor 647 goat anti-rabbit immunoglobulin G H&L (ab150079, 1:250 dilution; Abcam) for 3 hours. After the cells were washed, they were treated with ProLong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific). Fluorescence images were captured with BZ-X710 All-in-One Fluorescence Microscope (Keyence).
Statistical analysis
Statistical significance of differences between different groups was assessed with an unpaired 2-tailed Student t test. The survival of mice was analyzed by the Kaplan-Meier life test and by the log-rank test. P values < .05 were considered significant.
Results
CAMK2 inhibitor was identified by compound screening using MF-iPSCs
To maximize the advantages of MF-iPSCs and investigate the therapeutic targets for MF, we performed the compound screening, which has been quite limited in MF because of the paucity of humanized models. We established MF-iPSCs with integration-free episomal vectors from 3 different patients with MF harboring JAK2 V617F, CALR type 1, and CALR type 2 mutations (supplemental Table 1). CD34+ cells were collected from peripheral blood or BM. After stimulation with cytokines for 2 days, transduction with episomal vectors (OCT3/4, SOX3, KLF4, L-Myc, LIN28, sh-p53, and EBNA1) was performed (Figure 1A). One day after transduction, cells were reseeded onto mouse embryonic fibroblast (MEF) and cultured for 2 days. The medium was replaced with human ES media supplemented with 5 ng/mL basic fibroblast growth factor (bFGF). ES-like colonies were obtained, and the expression of stem cell markers, including SSEA-4 and TRA-1-60, was confirmed using immunofluorescence microscopy (Figure 1B). All MF-iPSCs developed teratoma with 3 germ layers, as confirmed via histological analyses (supplemental Figure 1A). We selected clones with the same driver mutation and karyotype as the parental cells (supplemental Figure 1B-C). Through hematopoietic differentiation, a population of cells coexpressing CD34 and CD43 was observed, indicative of hematopoietic progenitor formation (Figure 1C). In these cells, phosphorylation of STAT5 was enhanced in MF primary cells.23 We next assessed whether MF-HPCs differentiated from MF-iPSCs recapitulated the drug sensitivity of the original MF cells. Two compounds that were reported to be effective in MF samples were used for validation. Consistent with previous reports, JAK2 inhibitor (INCB18424, ruxolitinib) and heat shock protein 90 inhibitor (PU-H71) suppressed the viability of these cells (Figure 1D-E).24 Based on these results, newly established integration-free MF-HPCs were thought to recapitulate drug sensitivity, and this screening platform using the ATP luciferase assay was suitable for compound screening. The Screening Committee of Anticancer Drugs provided a list of 188 anticancer drugs and inhibitors (supplemental Table 4). For each compound, scores were assigned according to the results of the ATP luciferase assay, 2 (MF-HPCs were more sensitive at all 3 concentrations than normal HPCs), 1 (MF-HPCs were more sensitive at 2 sequential concentrations than normal HPCs), or 0 (the others). Seventeen compounds were assigned a score of 2 in the first screening using MF-HPCs with JAK2 V617F. In the second screening using MF-HPCs with mutant CALR type 1, KN-93 (CAMK 2 inhibitor) and aminoguanidine (nitric oxide synthase inhibitor) had scores of 2 and 1, respectively (Figure 2A-B). After the third screening using MF-HPCs having mutant CALR type 2, we identified KN-93 as a compound that inhibited the viability of MF-HPCs from all 3 patients, compared with normal HPCs (Figure 2C). For validation, we used trifluoperazine (TFP), another CAMK2 inhibitor, and confirmed that TFP had a similar inhibitory effect on MF-HPCs, indicating that CAMK2 inhibitors would be effective against MF cells harboring JAK2 V617F, CALR type 1, and CALR type 2 mutations (Figure 2D).
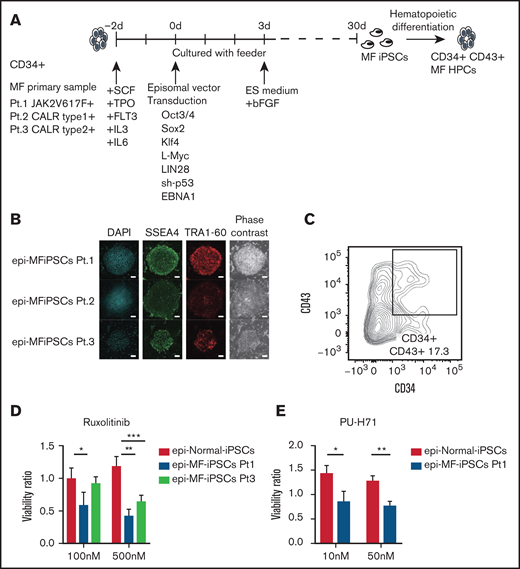
MF-iPSCs were established using episomal vectors. (A) Experimental scheme for generating iPSCs from MF patient samples is shown. MF-iPSCs with integration-free episomal vectors from 3 different MF patients harboring JAK2 V617F, CALR type 1, and CALR type 2 mutations were established. (B) Expression of pluripotency markers including SSEA-4 and TRA-1-60 was confirmed by immunostaining. Scale bar, 200 μm. (C) Cells differentiated from MF-iPSCs were analyzed by flow cytometry for the presence of CD34+ and CD43+ hematopoietic cells. (D) JAK2 inhibitor (ruxolitinib) was used for validation of drug sensitivity of MF-HPCs harboring JAK2 V617F and CALR type 2 mutation. Results are means ± standard deviation (SD). N = 3, independent experiments. Analysis of variance (ANOVA) test: *P < .05, **P < .01, ***P < .001. (E) HSP.90 inhibitor (PU-H71) was used for validation of drug sensitivity of MF-HPCs harboring JAK2 V617F. Results are means ± SD. N = 2, independent experiments. Unpaired 2-tailed Student t test: *P < .05, **P < .01.
MF-iPSCs were established using episomal vectors. (A) Experimental scheme for generating iPSCs from MF patient samples is shown. MF-iPSCs with integration-free episomal vectors from 3 different MF patients harboring JAK2 V617F, CALR type 1, and CALR type 2 mutations were established. (B) Expression of pluripotency markers including SSEA-4 and TRA-1-60 was confirmed by immunostaining. Scale bar, 200 μm. (C) Cells differentiated from MF-iPSCs were analyzed by flow cytometry for the presence of CD34+ and CD43+ hematopoietic cells. (D) JAK2 inhibitor (ruxolitinib) was used for validation of drug sensitivity of MF-HPCs harboring JAK2 V617F and CALR type 2 mutation. Results are means ± standard deviation (SD). N = 3, independent experiments. Analysis of variance (ANOVA) test: *P < .05, **P < .01, ***P < .001. (E) HSP.90 inhibitor (PU-H71) was used for validation of drug sensitivity of MF-HPCs harboring JAK2 V617F. Results are means ± SD. N = 2, independent experiments. Unpaired 2-tailed Student t test: *P < .05, **P < .01.
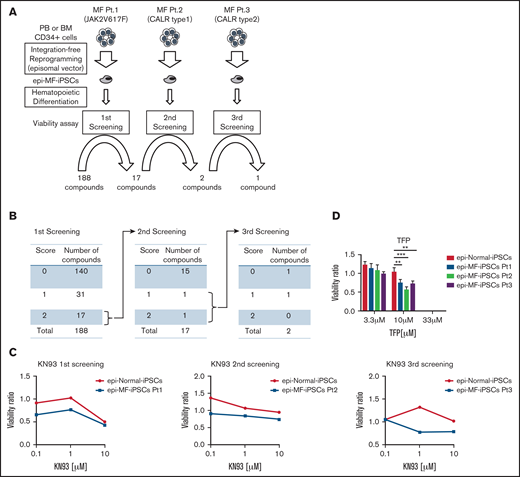
CAMK2 inhibitors were identified by the compound screening using MF-iPS cells established with episomal vectors. (A) Three rounds of the compound screening were performed with 188 compounds, including kinase inhibitors, genotoxic agents, and neurotransmitter inhibitors at 3 different concentrations. (B) The first screening was performed with MF-iPSCs harboring JAK2 V617F. After choosing 17compounds, the second screening was performed with MF-iPSCs harboring CALR type 1 mutation. After 3 rounds of screening, KN-93 (CAMK2 inhibitor) was identified as a therapeutic compound. (C) Results of the screening of KN-93 for all 3 MF-IPSCs are shown. (D) TFP, another CAMK2 inhibitor, was used to validate the effectiveness against all 3 MF-iPSCs. Results are means ± SD. ANOVA test: *P < .05, **P < .01, ***P < .001.
CAMK2 inhibitors were identified by the compound screening using MF-iPS cells established with episomal vectors. (A) Three rounds of the compound screening were performed with 188 compounds, including kinase inhibitors, genotoxic agents, and neurotransmitter inhibitors at 3 different concentrations. (B) The first screening was performed with MF-iPSCs harboring JAK2 V617F. After choosing 17compounds, the second screening was performed with MF-iPSCs harboring CALR type 1 mutation. After 3 rounds of screening, KN-93 (CAMK2 inhibitor) was identified as a therapeutic compound. (C) Results of the screening of KN-93 for all 3 MF-IPSCs are shown. (D) TFP, another CAMK2 inhibitor, was used to validate the effectiveness against all 3 MF-iPSCs. Results are means ± SD. ANOVA test: *P < .05, **P < .01, ***P < .001.
CAMK2 inhibition is effective in JAK2 inhibitor-sensitive cells and overcomes the resistance against JAK2 inhibitors
To address the efficacy of CAMK2 inhibition in other models of MF, we used BaF/3 cells expressing MPL W515L (MPLmu_S; IC50 of ruxolitinib = 112 nM). Consistent with the screening results, KN93 and TFP inhibited the proliferation of these cells (Figure 3A).
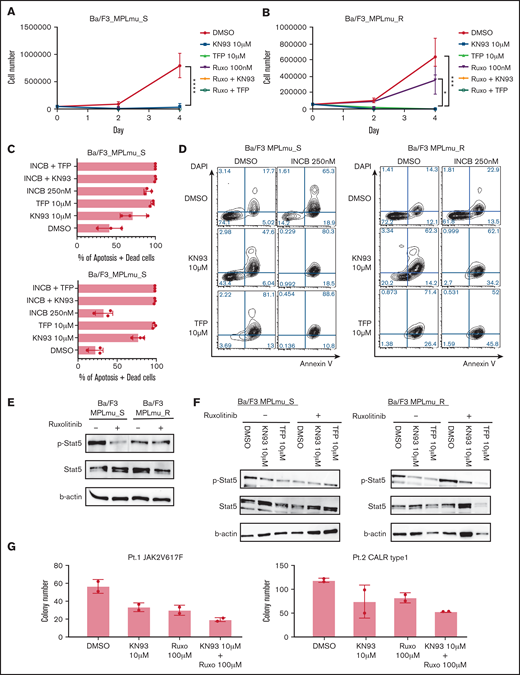
Effectiveness of CAMK2 inhibition in MF model cells. (A) Growth curve of BaF/3_MPLmu_S cells treated with DMSO, CAMK2 inhibitors (KN93 and TFP), ruxolitinib, and ruxolitinib in combination with CAMK2 inhibitors. Results are means ± SD. N = 3, independent experiments. ANOVA test: ****P < .0001. (B) BaF/3 cells expressing MPL W515L become resistant against JAK2 inhibitor 1 month after the exposure. Growth curve of BaF/3_MPLmu_R cells treated with DMSO, CAMK2 inhibitors (KN93 and TFP), and ruxolitinib and ruxolitinib in combination with CAMK2 inhibitors is shown. Results are means ± SD. N = 3, independent experiments. ANOVA test: *P < .05, ***P < .001. (C-D) Apoptosis of BaF/3_MPLmu_S cells and BaF/3_MPLmu_R cells was measured by Annexin V staining and FACS analysis when treated with INCB, KN93, TFP, or both. Results are means ± SD. N = 3, independent experiments. (E) BaF/3_MPLmu_S cells and BaF/3_MPLmu_R cells were examined by immunoblot for phosphorylation of STAT5 when treated with ruxolitinib. β-Actin was used as a loading control. (F) BaF/3_MPLmu_S cells and BaF/3_MPLmu_R cells were examined by immunoblot for phosphor-STAT5 when treated with ruxolitinib, CAMK2 inhibitor, or both. β-Actin was used as a loading control. (G) Colony-forming cell assay was performed with CD34+ cells from MF patients harboring CALR type 1 mutation and JAK2 V617F in the presence of KN93, ruxolitinib, or both. Results are means ± SD. N = 2, experimental replicates.
Effectiveness of CAMK2 inhibition in MF model cells. (A) Growth curve of BaF/3_MPLmu_S cells treated with DMSO, CAMK2 inhibitors (KN93 and TFP), ruxolitinib, and ruxolitinib in combination with CAMK2 inhibitors. Results are means ± SD. N = 3, independent experiments. ANOVA test: ****P < .0001. (B) BaF/3 cells expressing MPL W515L become resistant against JAK2 inhibitor 1 month after the exposure. Growth curve of BaF/3_MPLmu_R cells treated with DMSO, CAMK2 inhibitors (KN93 and TFP), and ruxolitinib and ruxolitinib in combination with CAMK2 inhibitors is shown. Results are means ± SD. N = 3, independent experiments. ANOVA test: *P < .05, ***P < .001. (C-D) Apoptosis of BaF/3_MPLmu_S cells and BaF/3_MPLmu_R cells was measured by Annexin V staining and FACS analysis when treated with INCB, KN93, TFP, or both. Results are means ± SD. N = 3, independent experiments. (E) BaF/3_MPLmu_S cells and BaF/3_MPLmu_R cells were examined by immunoblot for phosphorylation of STAT5 when treated with ruxolitinib. β-Actin was used as a loading control. (F) BaF/3_MPLmu_S cells and BaF/3_MPLmu_R cells were examined by immunoblot for phosphor-STAT5 when treated with ruxolitinib, CAMK2 inhibitor, or both. β-Actin was used as a loading control. (G) Colony-forming cell assay was performed with CD34+ cells from MF patients harboring CALR type 1 mutation and JAK2 V617F in the presence of KN93, ruxolitinib, or both. Results are means ± SD. N = 2, experimental replicates.
Long-term exposure to JAK2 inhibitors results in drug resistance.11 Hence, we tested the therapeutic efficacy of CAMK2G inhibitors in MF cells resistant to ruxolitinib (INCB18424). We cultured Ba/F3_MPLmu_S with ruxolitinib for 4 weeks and established ruxolitinib-resistant Ba/F3_MPLmu (MPLmu_R; IC50 of ruxolitinib = 642 nM). CAMK2 inhibitors alone or in combination with ruxolitinib dramatically decreased the growth of MPLmu_R cells (Figure 3B). The annexin V apoptosis assay revealed that both ruxolitinib and CAMK2 inhibitors induced apoptosis in MPLmu_S. Both KN93 and TFP induced apoptosis of MPLmu_R, whereas MPLmu_R cells were resistant to ruxolitinib-induced apoptosis (Figure 3C-D). Next, we explored the mechanism by which CAMK2 inhibitors inhibited the proliferation of these cells. Ruxolitinib decreased the phosphorylation of JAK2 and STAT5 in MPLmu_S, whereas persistent phosphorylation was observed in MPLmu_R (Figure 3E). This is consistent with the previous finding that resistance is caused by heterodimeric JAK-STAT activation that causes persistent phosphorylation of JAK2.11 In MPLmu_S, phosphorylation of STAT5 was substantially decreased in accordance with the suppression of proliferation when treated with CAMK2G inhibitors. In MPLmu_R, CAMK2G inhibitors decreased the phosphorylation of STAT5. The combination of CAMK2 inhibitors and ruxolitinib showed the decreased phosphorylation of STAT5 (Figure 3F). To address the effectiveness of CAMK2 inhibition in primary samples, we used CD34+ cells isolated from the peripheral blood mononuclear cells of patients with MF. KN93 or ruxolitinib alone inhibited the colony-forming capacity (CFC) of primary MF cells, and the combination of these showed an additive effect (Figure 3G).
CAMK2 γ plays a critical role in MF and demonstration of efficacy of CAMK2G inhibition in vitro and in vivo
CAMK2 has 4 subtypes, α, β, γ, and δ. Little is known about the functional and pathological roles of CAMK2 in MF. To determine the critical subtype of CAMK2 in MF, we assessed the expression of all CaM-kinase families and subtypes. We used 32D cells ectopically expressing wild-type MPL or MPL W515L (32D_MPLwt, 32D_MPLmu) and Ba/F3 cells ectopically expressing wild-type MPL or MPL W515L (Ba/F3_MPLwt, Ba/F3_MPLmu). In these cells, the CAMK2 γ subtype (CAMK2G) was exclusively expressed among all CAMK2 subtypes as revealed using qPCR (Figure 4A). As expected, CAMK2G protein was detected in Ba/F3_MPLwt and Ba/F3_MPLmu as observed using western blotting (Figure 4B). Next, we evaluated the efficacy of CAMK2G inhibition in vitro. Knockdown of CAMK2G by shRNA decreased IL-3–independent growth of Ba/F3_MPLmu (Figure 4C-D). These results suggest that CAMK2G plays a central role in the survival of these cells and that its inhibition decreases their proliferation. To assess the effect of CAMK2G inhibition in primary BM cells ectopically expressing MPL W515L, we used Camk2g knockout mice (Figure 4E; supplemental Figure 2A). Camk2g wt/− c-kit+ cells ectopically expressing MPL W515L tended to decrease CFC in the first round of replating and significantly decreased CFC in the second round of replating (Figure 4F). We also demonstrated that Camk2g wt/− c-kit+ cells ectopically expressing mock or wild-type MPL did not decrease CFC compared with Camk2g wt/wt c-kit+ cells (supplemental Figure 2B). We also used a specific CAMK2G inhibitor called berbamine to confirm the efficacy of CAMK2G inhibition.25,26 Berbamine decreased the expression of CAMK2G and inhibited CFC of c-kit+ BM cells from mice ectopically expressing MPL W515L in a dose-dependent manner, whereas berbamine did not affect the CFC of cells expressing wild-type MPL (Figure 4G-I). These results indicate that CAMK2G plays a critical role in the survival of MF cells. Furthermore, CAMK2G inhibition did not decrease the CFC of cells expressing wild-type MPL, suggesting that CAMK2G inhibition could be a safe therapeutic strategy of MF.
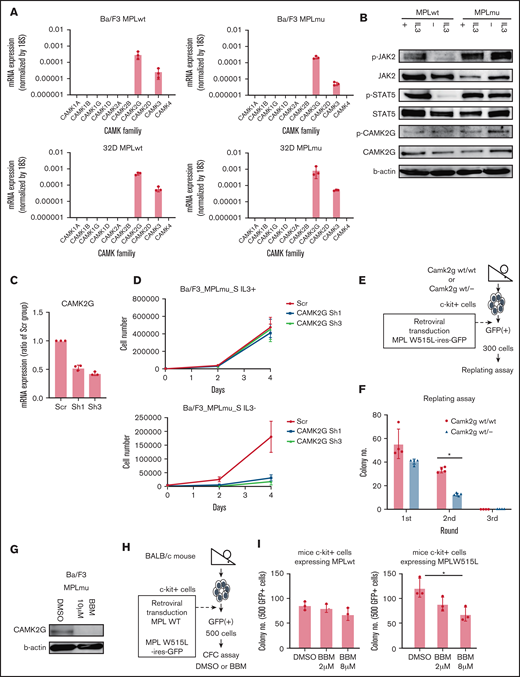
Identification of the critical subtype of CAMK2 in MF. (A) Gene expressions of CAMKs including CAMK1A, 1B, 1G, 1D, 2A, 2B, 2G, 2D, 3, and 4 were assessed by qPCR in BaF/3 and 32D cells overexpressing wild-type MPL and MPL W515L. (B) Immunoblot analysis of JAK, STAT5, CAMK2G, phospho-JAK, phospho-STAT5, and phosphor-CAMK2G. β-Actin was used as a loading control. (C) qPCR data of CAMK2G messenger RNA (mRNA) in BaF/3 cells overexpressing wild-type MPL and MPL W515L transduced with retroviruses carrying vectors with shRNAs or empty vector. Results are means ± SD. N = 3, independent experiments. (D) Proliferation curves of BaF/3 cells overexpressing wild-type MPL and MPL W515L transduced with retroviruses carrying vectors with shRNAs or empty vector. Results are means ± SD. N = 3, independent experiments. (E) Scheme of replating assay using Camk2g wt/− mouse BM. (F) The result of the colony-forming cell capacity of c-kit+ cells of wild and Camk2g knockout mice overexpressing MPL W515L. Results are means ± SD. N = 4, independent experiments. (G) Immunoblot analysis of pCAMK2G after treatment with berbamine (CAMK2G inhibitor) in BaF/3_MPLmu. β-Actin was used as a loading control. (H) Scheme of colony-forming cell assay with berbamine treatment. (I) Colony-forming cell assay was performed with BM cells overexpressing wild-type MPL and MPL W515L in the presence of DMSO or berbamine. Results are means ± SD. N = 3, independent experiments. ANOVA test: *P < .05.
Identification of the critical subtype of CAMK2 in MF. (A) Gene expressions of CAMKs including CAMK1A, 1B, 1G, 1D, 2A, 2B, 2G, 2D, 3, and 4 were assessed by qPCR in BaF/3 and 32D cells overexpressing wild-type MPL and MPL W515L. (B) Immunoblot analysis of JAK, STAT5, CAMK2G, phospho-JAK, phospho-STAT5, and phosphor-CAMK2G. β-Actin was used as a loading control. (C) qPCR data of CAMK2G messenger RNA (mRNA) in BaF/3 cells overexpressing wild-type MPL and MPL W515L transduced with retroviruses carrying vectors with shRNAs or empty vector. Results are means ± SD. N = 3, independent experiments. (D) Proliferation curves of BaF/3 cells overexpressing wild-type MPL and MPL W515L transduced with retroviruses carrying vectors with shRNAs or empty vector. Results are means ± SD. N = 3, independent experiments. (E) Scheme of replating assay using Camk2g wt/− mouse BM. (F) The result of the colony-forming cell capacity of c-kit+ cells of wild and Camk2g knockout mice overexpressing MPL W515L. Results are means ± SD. N = 4, independent experiments. (G) Immunoblot analysis of pCAMK2G after treatment with berbamine (CAMK2G inhibitor) in BaF/3_MPLmu. β-Actin was used as a loading control. (H) Scheme of colony-forming cell assay with berbamine treatment. (I) Colony-forming cell assay was performed with BM cells overexpressing wild-type MPL and MPL W515L in the presence of DMSO or berbamine. Results are means ± SD. N = 3, independent experiments. ANOVA test: *P < .05.
To test the efficacy of CAMK2G inhibition in vivo, we transplanted GFP+ c-kit+ cells from the BM ectopically expressing MPL W515L into irradiated BALB/c mice. These mice develop a fatal hematologic disease characterized by peripheral blood leukocytosis and hepatosplenomegaly.27 The recipient mice were divided into 3 groups and treated with berbamine via daily intraperitoneal administration at the following doses: 0 (administered PBS only), 50, or 100 mg/kg, from day 14 to day 23 (Figure 5A). In the PBS group, all animals died because of disease progression by day 40. In contrast, berbamine at both doses was effective in treating MPL W515L–induced disease, as there was a significant prolongation of survival (P < .05) (Figure 5B). We also assessed the effects of berbamine on MPL W515L–induced leukocytosis, anemia, thrombocytosis, and extramedullary hematopoiesis. There were no differences in WBC counts before treatment (supplemental Figure 3A). Mice receiving berbamine (50 and 100 mg/kg) showed a significant reduction in WBC counts compared with mice treated with PBS (P < .05). Hemoglobin levels and platelet counts were not significantly different between the groups on day 24 (Figure 5C). Mice receiving 100 mg/kg berbamine had a significant reduction in spleen length, whereas there were no differences in spleen, liver, or body weights between groups on day 24 (Figure 5D; supplemental Figure 3B-C). To clarify the mechanism by which berbamine affects MF model mice in vivo, we performed an apoptosis assay in peripheral blood. We found that berbamine treatment induced apoptosis in MF model cells in vivo (Figure 5E-F). These findings indicate that CAMK2G is a potential therapeutic target for MF.
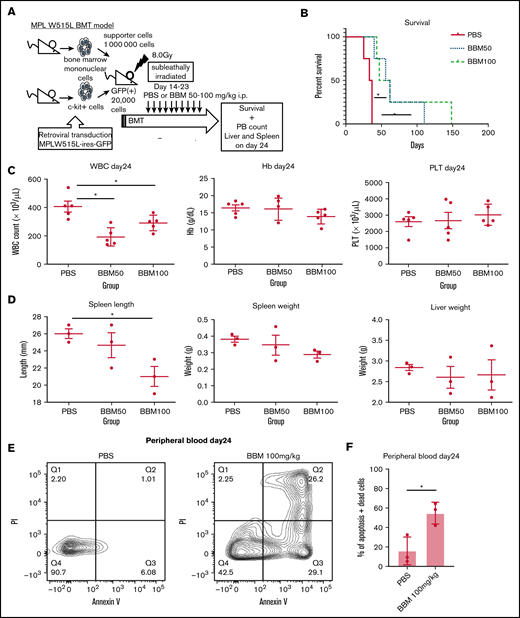
Confirmation of the effectiveness of CAMK2G inhibition in vivo using retroviral BM transplantation model. (A) Sublethally irradiated BALB/c mice were transplanted with c-kit+ BM cells transduced with retroviruses carrying the vector of MPL W515L. These mice were treated with intraperitoneal injections of berbamine (BBM) from day 14 to 23. (B) The survival curves for bone marrow transplantation (BMT) model mice treated with berbamine. Log-rank test: *P < .05, N = 4 mice per group. (C) Parameters, including white blood cell count (WBC count), hemoglobin (Hb), and platelet count (PLT), are shown. N = 5 mice per group. (D) Spleen length, spleen weight, and liver weight are shown N = 3 mice per group. ANOVA test: *P < .05. (E-F) The result of the apoptosis assay in peripheral blood cells of MPL W515L-induced MF model mice under BBM treatment. Peripheral blood cells were measured by FACS after Annexin V + propidium iodide (PI) staining on day 24 with or without BBM treatment. Results are means ± SD. N = 3 mice per group. Unpaired 2-tailed Student t test: *P < .05.
Confirmation of the effectiveness of CAMK2G inhibition in vivo using retroviral BM transplantation model. (A) Sublethally irradiated BALB/c mice were transplanted with c-kit+ BM cells transduced with retroviruses carrying the vector of MPL W515L. These mice were treated with intraperitoneal injections of berbamine (BBM) from day 14 to 23. (B) The survival curves for bone marrow transplantation (BMT) model mice treated with berbamine. Log-rank test: *P < .05, N = 4 mice per group. (C) Parameters, including white blood cell count (WBC count), hemoglobin (Hb), and platelet count (PLT), are shown. N = 5 mice per group. (D) Spleen length, spleen weight, and liver weight are shown N = 3 mice per group. ANOVA test: *P < .05. (E-F) The result of the apoptosis assay in peripheral blood cells of MPL W515L-induced MF model mice under BBM treatment. Peripheral blood cells were measured by FACS after Annexin V + propidium iodide (PI) staining on day 24 with or without BBM treatment. Results are means ± SD. N = 3 mice per group. Unpaired 2-tailed Student t test: *P < .05.
CAMK2G is phosphorylated by MPL signal via JAK2
Next, we sought to elucidate the molecular mechanism of CAMK2G in the pathogenesis of MF. We investigated the potential interactions between the MPL-JAK2 pathway and CAMK2G. To elucidate the signaling pathway related to CAMK2G, we constructed 32D cells expressing both wild-type and mutant MPL together with CAMK2G. The phosphorylation of CAMK2G was enhanced when cells were stimulated by TPO (10 ng/mL), indicating that CAMK2G is located downstream of the MPL (Figure 6A-B). Ruxolitinib (250 nM) suppressed the phosphorylation of CAMK2G in 32D cells expressing both CAMK2G and wild-type MPL or W515L in the presence of IL-3 (Figure 6C). Besides, knockdown of JAK2 in 32D cell line coexpressing both MPL W515L and CAMK2G suppressed the phosphorylation of CAMK2G (Figure 6D). Next, we rescued wild-type JAK2 in these cells. The expression of JAK2 was significantly higher in cells transduced with a wild-type JAK2 vector, as confirmed by western blotting (supplemental Figure 4B). Enforced expression of wild-type JAK2 increased the phosphorylation of CAMK2G in cells transduced with a shJAK2-2 vector (Figure 6E). These results suggest that both wild-type and mutant MPL signals phosphorylate the CAMK2G protein via JAK2. To evaluate the status of endogenous CAMK2G, we examined the localization of CAMK2G. A previous study reported that CAMK2G translocates into the nucleus upon phosphorylation.28 Consistent with this, we observed that CAMK2G in Ba/F3 expressing MPL W515L mutant was localized more in the nucleus than in those expressing wild-type MPL in the absence of IL-3 (Figure 6F). This finding suggests that CAMK2G is potentially involved in promoting MF by altering of gene expression profiles. We also assessed the signaling pathway using Ba/F3_MPLmu cells treated with berbamine. Berbamine promptly suppressed CAMK2G and STAT5 and did not affect the phosphorylation of JAK2 (Figure 6G), indicating that STAT5 is one of the downstream targets of berbamine. Taken together, these results indicate that CAMK2G is an effector downstream of the MPL-JAK2 signal and STAT5 is one of the effectors downstream of CAMK2G.
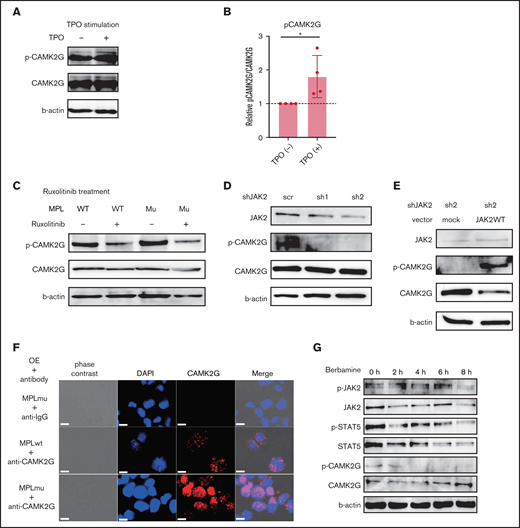
CAMK2G is phosphorylated by MPL signal via JAK2. (A) Immunoblotting of phospho-CAMK2G in 32D cells overexpressing wild-type MPL together with CAMK2G when cells were stimulated by TPO in the absence of IL-3. β-Actin was used as a loading control. (B) The result of the immunoblotting is quantified. The ratio of phospho-CAMK2G/CAMK2G is shown. N = 4, independent experiments, unpaired 2-tailed Student t test: *P < .05. (C) Immunoblotting of phospho-CAMK2G in 32D cells overexpressing wild-type MPL or MPL W515L together with CAMK2G when cells were treated with ruxolitinib. β-Actin was used as a loading control. (D) Immunoblotting of phospho-CAMK2G in 32D cells overexpressing MPL W515L and CAMK2G after knockdown of JAK2 with shRNA. β-Actin was used as a loading control. (E) Immunoblotting of phospho-CAMK2G in JAK2 knockdown cells after cells were transduced with empty or wild-type JAK2 vectors. β-Actin was used as a loading control. (F) Immunostaining using anti-CAMK2G antibody was performed in Ba/F3_MPLwt and Ba/F3_MPLmu. Scale bar, 10 μm. (G) Immunoblotting of phospho-JAK2, phosphor-STAT5 in Ba/F3_MPLmu when treated with berbamine. β-Actin was used as a loading control.
CAMK2G is phosphorylated by MPL signal via JAK2. (A) Immunoblotting of phospho-CAMK2G in 32D cells overexpressing wild-type MPL together with CAMK2G when cells were stimulated by TPO in the absence of IL-3. β-Actin was used as a loading control. (B) The result of the immunoblotting is quantified. The ratio of phospho-CAMK2G/CAMK2G is shown. N = 4, independent experiments, unpaired 2-tailed Student t test: *P < .05. (C) Immunoblotting of phospho-CAMK2G in 32D cells overexpressing wild-type MPL or MPL W515L together with CAMK2G when cells were treated with ruxolitinib. β-Actin was used as a loading control. (D) Immunoblotting of phospho-CAMK2G in 32D cells overexpressing MPL W515L and CAMK2G after knockdown of JAK2 with shRNA. β-Actin was used as a loading control. (E) Immunoblotting of phospho-CAMK2G in JAK2 knockdown cells after cells were transduced with empty or wild-type JAK2 vectors. β-Actin was used as a loading control. (F) Immunostaining using anti-CAMK2G antibody was performed in Ba/F3_MPLwt and Ba/F3_MPLmu. Scale bar, 10 μm. (G) Immunoblotting of phospho-JAK2, phosphor-STAT5 in Ba/F3_MPLmu when treated with berbamine. β-Actin was used as a loading control.
Discussion
In this study, we established MF iPSCs that recapitulate the disease phenotype and used these cells to screen compounds. We found that CAMK2 inhibitor can be a promising drug for MF, and that CAMK2G inhibition is effective in MF model cells and mice. The results of this study also indicate that CAMK2G is a critical downstream effector of the MPL-JAK2 signaling pathway.
The establishment of iPSCs derived from patient samples has been reported and can be a promising strategy to elucidate the pathogenesis of the disease and identified new therapeutic strategy.14,15,29 Although we previously established iPSCs derived from MF patient samples using retroviral vectors, this method can cause some unintentional effects by modifying the host genes.17 Here, we established iPSCs derived from MF patient samples with integration-free episomal vectors to minimize the bias caused in the reprogramming process and confirmed that these cells recapitulated the disease phenotype and drug sensitivity. MF-iPSCs can be a new humanized disease model, and a large number of patient-derived cells can be obtained using this method for further analyses. By performing compound screening with MF-iPSCs, CAMK2G was identified as a new therapeutic target for MF. It is known that CAMK2G is highly expressed in brain and plays a role in synaptic plasticity and memory formation.28,30 There are a few studies on the pathological roles of CAMK2G in cancer. It has been reported that CAMK2G plays a role in the proliferation of leukemic cells and blast crisis in chronic myeloid leukemia.31,32 It has also been reported that CAMK2G promotes invasion and migration in breast cancer and enhances tumorigenicity in lung cancer.33,34 However, the functional and pathological roles of CAMK2G in MF have not yet been investigated. We show here that inhibition of CAMK2G decreased the growth of MF cells in vitro, prolonged survival, and ameliorated splenomegaly in MF model mice in vivo. Berbamine is isolated from the traditional Chinese herbal medicine Berberis amurensis and possesses a unique profile of biological activity, including anti-inflammatory and antitumor activities.26,35-38 Although berbamine specifically inhibits CAMK2G by binding to the ATP-binding pocket, it is also known to be a potent calcium channel blocker, an inhibitor of the NF-kB pathway and BCR-ABL fusion gene.26,35,37-39 As such, berbamine can be a treatment option; however, further research is required to identify more effective drugs targeting CAMK2G. JAK2 inhibitors, including ruxolitinib, can ameliorate several symptoms of MF, whereas they cannot eliminate MF clones in most patients. Patients need to continue the treatment to control MF, and the long-term treatment with JAK2 inhibitors often leads to drug resistance.11 CAMK2G inhibition decreases the growth of the MF model cells resistant to JAK2 inhibitors. The results of the present study indicate CAMK2G inhibition can be a new therapeutic strategy for MF that can overcome drug resistance against JAK2 inhibitors. In our study, CAMK2G inhibition did not affect the CFC of murine BM cells expressing wild-type MPL. Although complete deletion of JAK2 causes lethal anemia and thrombocytopenia in mice, CAMK2G knockout mice did not show any hematopoietic defects.18,25,40 These facts support the idea that CAMK2G is important exclusively in MF cells and that its inhibition is a promising therapeutic strategy with few or no hematological side effects. Because we demonstrated that the combination of JAK2 inhibitor and CAMK2 inhibitors showed additive effects, a combination of these agents may be useful in clinical settings. Moreover, we can achieve efficacy with lower doses and fewer dose-dependent side effects of each agent. This strategy might be useful to more eligible patients for therapies employing these drugs. We also showed that CAMK2G is located downstream of the MPL-JAK2 signaling pathway, which plays important roles in MF. This result is consistent with our finding that CAMK2G inhibition is effective against resistant MF cells. Resistance against JAK2 inhibitors is associated with the reactivation of JAK-STAT signaling by heterodimerization between activated JAK2 and JAK1/TYK2.11 CAMK2G inhibition decreases the activated signaling from JAK2. CAMK2G is associated with several signal transductions, including MAPK, JAK-STAT, glycogen synthase kinase 3β/β-catenin, and c-Myc pathways.25,41 Because CAMK2G inhibition with berbamine decreases phosphorylation of STAT5, STAT5 would be one of the downstream effectors of CAMK2G. Given that berbamine possesses a wide range of biological activities and limited specificity as described above, there may be other pathways related to MF in the downstream of CAMK2G. Further studies are required to understand how CAMK2G triggers BM fibrosis. Collectively, our data indicate that CAMK2G is involved in the MPL-JAK signaling pathway in MF and is a promising therapeutic target for overcoming drug resistance.
Acknowledgments
The authors thank T. Kitamura for Plat-E and Plat-A packaging cells, H. Nakauchi for pGCDNsam-IRES-EGFP retroviral vector, and K. Okita for pCXLE-hOCT3/4-shp53-F, pCXLE-hSK, pCXLE-hUL and pCXWB-EBNA1. They thank Yoshiko Ito, Yoko Hokama, and Keiko Tanaka for expert technical assistance. They also thank Eric Olson (University of Texas Southwestern Medical Center, Dallas, Texas) and Johannes Backs (University of Heidelberg, Heidelberg, Germany) for providing them with CAMK2G knockout mice.
This work was supported in part by Advanced Research and Development Programs for Medical Innovation (AMED-CREST), the Japan Society for the Promotion of Science (JSPS) KAKENHI 14J02953 and 16K19568 and a grant from Foundation for Promotion of Cancer Research, Okinaka Memorial Institute for Medical Research, Takeda Science Foundation, and the Tokyo Biochemical Research Foundation. S.A. has received research funding from Bristol-Myers Squibb.
Authorship
Contribution: M.M., K.S., and M.K. conceived and designed the study; M.M. and K.S. performed the experiments; H.M., K.T., Y.M., S.Y., S.A., and Y.K. assisted with the experimental performance; M.M. and K.S. wrote and modified the manuscript; and all authors gave intellectual input to the study and approved the final version of the manuscript.
Conflict-of-interest disclosure: M.K. reports grants from Pfizer Seiyaku KK, Astellas Pharma, Kyowa Hakko Kirin Co, Ltd, Takeda Pharmaceutical Company Ltd, Ono Pharmaceutical Co, Ltd, Nippon Shinyaku Co, Ltd, Bristol-Meyer Squib, Novartis Pharmaceuticals, other from Shionogi & Co, Ltd, and other from Celgene KK, outside of the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Mineo Kurokawa, Department of Hematology and Oncology, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-City Tokyo, 113-8655 Japan; e-mail: kurokawa-tky@umin.ac.jp.

