

Key Points
Donor-derived, tumor-associated, antigen-specific T cells targeting survivin, PRAME, and WT1 are safe in acute leukemia patients post-BMT.
One-year OS in patients who relapsed early post-BMT was 42.8% postrelapse, whereas 88.9% of evaluable high-risk patients were alive at 1 year.

Abstract
Patients with hematologic malignancies relapsing after allogeneic blood or marrow transplantation (BMT) have limited response to conventional salvage therapies, with an expected 1-year overall survival (OS) of <20%. We evaluated the safety and clinical outcomes following administration of a novel T-cell therapeutic targeting 3 tumor-associated antigens (TAA-T) in patients with acute leukemia who relapsed or were at high risk of relapse after allogeneic BMT. Lymphocytes obtained from the BMT donor were manufactured to target TAAs WT1, PRAME, and survivin, which are over-expressed and immunogenic in most hematologic malignancies. Patients received TAA-T infusions at doses of 0.5 to 4 × 107/m2. Twenty-three BMT recipients with relapsed/refractory (n = 11) and/or high-risk (n = 12) acute myeloid leukemia (n = 20) and acute lymphoblastic leukemia (n = 3) were infused posttransplant. No patient developed cytokine-release syndrome or neurotoxicity, and only 1 patient developed grade 3 graft-versus-host disease. Of the patients who relapsed post-BMT and received bridging therapy, the majority (n = 9/11) achieved complete hematologic remission before receiving TAA-T. Relapsed patients exhibited a 1-year OS of 36% and 1-year leukemia-free survival of 27.3% post–TAA-T. The poorest prognosis patients (relapsed <6 months after transplant) exhibited a 1-year OS of 42.8% postrelapse (n = 7). Median survival was not reached for high-risk patients who received preemptive TAA-T posttransplant (n = 12). Although as a phase 1 study, concomitant antileukemic therapy was allowed, TAA-T were safe and well tolerated, and sustained remissions in high-risk and relapsed patients were observed. Moreover, adoptively transferred TAA-T detected by T-cell receptor V-β sequencing persisted up to at least 1 year postinfusion. This trial was registered at clinicaltrials.gov as #NCT02203903.
Introduction
Relapse is the most frequent cause of death after allogeneic blood or marrow transplantation (BMT) for high-risk hematologic malignancies. The chance of relapse post-BMT for patients with acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL) ranges from 4% to 80%,1 with a <10% chance of remaining alive and disease-free at 1 year if relapse occurs within 6 months of BMT.2-5 Pediatric patients may fare better than adults, but 1-year overall survival (OS) following relapse within 6 months of BMT still remains limited at 20% to 25%.5,6 Current strategies to treat post-BMT relapse, such as chemotherapy and donor lymphocyte infusions (DLI) to enhance the immunologic graft-versus-leukemia (GVL) effect, have low efficacy and are associated with graft-versus-host disease (GVHD).1,7,8 Although second allo-transplants to reinstate GVL effects are occasionally effective in some patients who enter a remission, they are not effective in patients who relapse early after BMT.3,6,9,10 New immunotherapeutic strategies, notably transgenic chimeric antigen receptor (CAR) T cells, have shown promise in patients with ALL11,12 but can have significant toxicities and have shown minimal efficacy in AML to date. Furthermore, the shared expression of CAR-targeted AML surface antigens on normal myeloid precursors can incur myelotoxicity.13
An alternative strategy to boost the GVL effect is to infuse donor-derived T cells targeting tumor-associated antigen (TAA) peptides as circulating TAA-specific T cells (TAA-T) are associated with maintenance of remission post-BMT.14 In this phase 1 clinical trial, a novel, multitargeted T-cell therapeutic was administered in an attempt to decrease the likelihood of tumor evasion while broadening treatment options for BMT recipients with relapsed or high-risk acute leukemia regardless of HLA type. The TAAs WT1, survivin, and PRAME were selected as targets because they are widely overexpressed in relapsed and refractory hematopoietic malignancies.15-17 Here, we report on the safety, tolerability, persistence, and clinical effects of this novel TAA-T treatment as an approach to prolong survival of patients with high-risk or relapsed leukemia after BMT.
Methods
Study design
A prospective, open-label, pilot clinical trial (NCT02203903; IND16135) was conducted at Children’s National and Johns Hopkins Hospitals as a dose-escalation study with doses ranging from 0.5 × 107/m2 (dose level 1) to 4.0 × 107/m2 (dose level 4), with up to 7 infusions per patient depending on availability of the product. This study was approved by the institutional review board at each institution and was conducted in accordance with the principals of the Declaration of Helsinki and Good Clinical Practice guidelines. See supplemental Methods for full details including eligibility. Steroids exceeding 0.5 mg/kg per day prednisone equivalent at the time of TAA-T or investigational T-cell suppressive therapies in the prior 28 days were exclusion criteria. Patients with relapsed disease could receive cytotoxic therapies prior to and after TAA-T as deemed necessary.
TAA-T generation
TAA-T products were manufactured under Good Manufacturing Practice conditions from peripheral blood mononuclear cells (PBMCs) obtained from corresponding BMT donors as previously described18 ; see supplemental Methods. Briefly, donor-derived dendritic cells were pulsed with TAA peptide libraries and cocultured with donor-derived T cells in the presence of cytokines (IL-7, IL-15, IL-12, IL-6) and restimulated in the presence of IL-2.19-21 TAA-T products were validated for identity, phenotype, sterility, lack of alloreactivity, and cryopreserved prior to administration.
TAA-T immunophenotyping and functional analysis
TAA-T products were analyzed with monoclonal antibodies against CD3, CD4, CD8, CD127, CD14, CD19, CD25, CD16, CD56, TCRγδ, CD95, CD45RO, CD28, CCR7, and CD62L.
TAA-T product-specific activity against WT1, survivin, and PRAME pepmixes (JP Peptide Technology, Berlin, Germany) was evaluated as previously described using anti–interferon gamma (IFNγ) Enzyme-Linked Immunospot (ELISpot).18 Assays were read by Zellnet consulting (Fort Lee, NJ). TAA-T functionality was also assessed by flow cytometric detection of intracellular IFN-γ and tumor necrosis factor alpha (TNF-α) in response to WT1, PRAME, or survivin peptides, as described in supplemental Methods and as published.18 Cells were acquired on a Cytoflex S (Beckman Coulter, Brea, CA). Gating strategies are shown in supplemental Figures 1-4. Data analyses were performed using FlowJo version 10.5.
Cytotoxicity assay
The cytolytic function of the TAA-T product was evaluated against the HLA-A*02 AML cell line (THP-1) as described in supplemental Methods. Briefly, Cell Trace Violet–labeled THP-1 tumor cells were cocultured with TAA-T or nonspecific T cells (PBMC). Cocultures were labeled with monoclonal antibodies against CD3, CD45RO, and CD33, and the cytotoxicity of TAA-T products vs nonspecific T cells was assessed by lysis of CD33+Violet+ cells. The number of tumor cells remaining was reported as a proportion of the number of tumor cells plated. Tumor cells alone provided background viability. Cells were acquired on a Cytoflex S (Beckman Coulter). Data analysis was performed using FlowJo version 10.5.
Postinfusion monitoring
Patients were monitored for immediate infusion-related toxicities for 1 hour after each TAA-T infusion. Patients were subsequently evaluated weekly for 6 weeks for GVHD and other toxicities using the Common Terminology Criteria for Adverse Events version 4.03.
Clinical response definitions
Clinical responses after TAA-T were categorized as: continued complete remission (CCR; continued absence of morphologic or extramedullary disease); complete response (CR; absence of morphologic or extramedullary disease that is either minimal residual disease [MRD]-positive or -negative); partial response (PR; >10% decrease in leukemia disease without meeting CR criteria); mixed response (decreased evidence of disease in some sites but not all); progressive disease (PD; worsening of all areas of disease); or stable disease (changes insufficient to qualify for CR, PR, mixed response, or PD).
Immunosequencing
Samples of DNA isolated from the TAA-T product and recipient research blood (0.5 × 106 per sample) were sent to Adaptive Biotechnologies (Seattle, WA) for T-cell receptor V-β (TCRVbeta) sequencing on the Immunoseq platform with analysis and compilation of sequence results.
Statistical analysis
OS and leukemia-free survival (LFS) were calculated from the time of TAA-T infusion to death from any cause, except when otherwise indicated. Survival curves were estimated by Kaplan-Meier and described as 1-year OS, LFS, or as median survival for groups with follow-up <1 year. Curves were compared using the log-rank test. Paired Wilcoxon test was used to compare mean antigen responses with a cutoff P value for significance of <.05. No adjustments were made for multiple comparisons.
Results
Patient characteristics
Twenty-three patients with relapsed/refractory (n = 11) or high-risk (n = 12) AML (n = 20) and ALL (n = 3) were infused with a donor-derived TAA-T-cell product post-BMT. The median age at time of first TAA-T infusion was 33 years (range, 1-74), with 5 pediatric patients (<18 years). Eleven patients were female, and 12 were male. The donor was HLA matched for 11 patients and haploidentical-related in 12. All TAA-T products were manufactured from the patient’s BMT donor.
In the group with relapsed/refractory disease posttransplant (n = 11), the median time from first transplant to relapse was 5 months (range, 56-310 days) (Table 1). Notably, 9 of these patients were MRD+ (n = 2) and had hematologic relapse (n = 5) or extramedullary relapse (n = 2) within 6 months after transplant, emphasizing the poor prognosis of the patients in this study. All relapsed patients with hematologic and extramedullary relapse received at least 1 other therapy prior to TAA-T infusion, including 1 or more courses of chemotherapy (n = 9), second transplant (n = 2), DLI (n = 2), IFNα (n = 1), and/or TKI (n = 2). Patient P8 had persistent MRD with BCR/ABL positivity in the marrow following transplant and treatment with dasatinib and was considered refractory. Two patients (P9, P10) received their first TAA-T infusion after a second BMT. Patient P9 received a second BMT for relapse 10 months following his first transplant and achieved second complete remission prior to the first TAA-T infusion. Patient P10 developed MRD 180 days after her first BMT. She became MRD+ again 271 days after a second transplant. MRD was reduced to 0.01% with DLI and IFNα prior to receiving the first TAA-T infusion. Ultimately, most patients (9/11, 81.8%) achieved hematologic and extramedullary CR prior to TAA-T-cell infusion, whereas 3 additional patients remained MRD+ (P1, P8, P10).
Characteristics and disease response in patients with relapsed disease prior to TAA-T infusion.
| Patient ID | Age/ sex | Diagnosis indication for BMT | Donor and transplant type | Time to evidence of disease after transplant (days) | Status at relapse | Postrelapse treatment | Status at TAA-T cell infusion (evaluation timepoint preinfusion) | Day post-BMT at time of TAA-T infusion | ALC at first TAA-T cell infusion (k/IU) | TAA-T dose level (number of doses) | Best response postinfusion | Time to relapse postinfusion (days) | Survival postinfusion (days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 24 Years/M | B-ALL Ph+ | HLA = sibling TBI MA | 75 | CNS3 BM 15% blasts | Azacitidine,DLI, XRT, TKI, IT chemotherapy | BCR/ABL+, CNS− (34 d) | +896 | 0.27 | 1 (1) | SD | 21 | 28 |
| 2 | 64 Years/M | MDS/AML | HLA = sibling NMA | 155 | BM 27% blasts | HIDAC × 2 | CR (9 d) | +330 | 0.77 | 1 (1) | PD | 42 | 169 |
| 3 | 48 Years/F | B-ALL | HLA = sibling MA | 56 | Extramedullary leukemia | VP16/MT × 1, blinatumomab, Cytarabine | PD (3 d) | +423 | 2.21 | 2 (1) | SD | 19 | 255 |
| 4 | 54 Years/M | MDS/AML tri8,13,20 MLL RUNX1T1 | HLA = sibling NMA | 107 | BM 80% blasts | HIDAC × 2 | CR (17 d) | +184 | 0.37 | 2 (1) | PD | 36 | 137 |
| 5 | 68 Years/F | MDS/AML tri8, 5q-, mo7 | Haplo son NMA | 117 | BM 5% blasts | Azacitidine × 3 | CR (9 d) | +222 | 1.43 | 3 (3) | CCR | 167 | 422 |
| 6 | 70 Years/M | MDS/AML tri8, 14 | Haplo son NMA | 179 | BM 80% blasts | ACDVP16 × 1 | CR (53 d) | +283 | 0.86 | 3 (1) | PD | 53 | 95 |
| 7 | 58 Years/M | AML 9q- tri21, Mut CEBPa,Kit, IK2F | HLA = sibling NMA | 289 | BM 60% blasts | ACDVP16 × 1HIDAC ×2 | CR (123 d) | +455 | 1.81 | 3 (4) | CCR | 518 | 1150 |
| 8 | 9 Years/F | B-ALL Ph+ TP53 mutation | HLA = sibling MA | N/A | Persistent positive BCR/ABL | TKI | Persistent positive BCR/ABL, MRD− (15 d) | +231 | 2.82 | 4 (2) | CCR (MRD−, BCR/ABL undetectable) | N/A (MRD+ day 105) | 1160+ |
| 9 | 21 Years/M | AML GATA2 FLT3+ | Haplo mother NMA | No relapse after 2nd transplant (relapsed 310 d after 1st transplant) | Marrow morphology “suspicious” for AML | Second BMT, azacitidine | CR (28 d) | +167 | 1.09 | 4 (1) | CCR | N/A | 812+ |
| 10 | 4 Years/F | AML MLL rearrangement | Haplo father RI | 271 post–2nd transplant (relapsed 180 d after 1st transplant) | BM blasts 0.08% (0.48% 3 mo later) | IFNa, DLI | MRD 0.01% (6 d) | +460 | 2.12 | 4 (1) | PR | 90 | 323 |
| 11 | 61 Years/M | MDS/AML EBB1, p53 mutation | MiniHaplo son NMA | 115 | BM blasts 20% | Azacitidine (post–TAA-T cells) | Persistent blasts (20 d) | +135 | 0.71 | 4 (1) | SD | 64 (PD) | 150 |
| Patient ID | Age/ sex | Diagnosis indication for BMT | Donor and transplant type | Time to evidence of disease after transplant (days) | Status at relapse | Postrelapse treatment | Status at TAA-T cell infusion (evaluation timepoint preinfusion) | Day post-BMT at time of TAA-T infusion | ALC at first TAA-T cell infusion (k/IU) | TAA-T dose level (number of doses) | Best response postinfusion | Time to relapse postinfusion (days) | Survival postinfusion (days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 24 Years/M | B-ALL Ph+ | HLA = sibling TBI MA | 75 | CNS3 BM 15% blasts | Azacitidine,DLI, XRT, TKI, IT chemotherapy | BCR/ABL+, CNS− (34 d) | +896 | 0.27 | 1 (1) | SD | 21 | 28 |
| 2 | 64 Years/M | MDS/AML | HLA = sibling NMA | 155 | BM 27% blasts | HIDAC × 2 | CR (9 d) | +330 | 0.77 | 1 (1) | PD | 42 | 169 |
| 3 | 48 Years/F | B-ALL | HLA = sibling MA | 56 | Extramedullary leukemia | VP16/MT × 1, blinatumomab, Cytarabine | PD (3 d) | +423 | 2.21 | 2 (1) | SD | 19 | 255 |
| 4 | 54 Years/M | MDS/AML tri8,13,20 MLL RUNX1T1 | HLA = sibling NMA | 107 | BM 80% blasts | HIDAC × 2 | CR (17 d) | +184 | 0.37 | 2 (1) | PD | 36 | 137 |
| 5 | 68 Years/F | MDS/AML tri8, 5q-, mo7 | Haplo son NMA | 117 | BM 5% blasts | Azacitidine × 3 | CR (9 d) | +222 | 1.43 | 3 (3) | CCR | 167 | 422 |
| 6 | 70 Years/M | MDS/AML tri8, 14 | Haplo son NMA | 179 | BM 80% blasts | ACDVP16 × 1 | CR (53 d) | +283 | 0.86 | 3 (1) | PD | 53 | 95 |
| 7 | 58 Years/M | AML 9q- tri21, Mut CEBPa,Kit, IK2F | HLA = sibling NMA | 289 | BM 60% blasts | ACDVP16 × 1HIDAC ×2 | CR (123 d) | +455 | 1.81 | 3 (4) | CCR | 518 | 1150 |
| 8 | 9 Years/F | B-ALL Ph+ TP53 mutation | HLA = sibling MA | N/A | Persistent positive BCR/ABL | TKI | Persistent positive BCR/ABL, MRD− (15 d) | +231 | 2.82 | 4 (2) | CCR (MRD−, BCR/ABL undetectable) | N/A (MRD+ day 105) | 1160+ |
| 9 | 21 Years/M | AML GATA2 FLT3+ | Haplo mother NMA | No relapse after 2nd transplant (relapsed 310 d after 1st transplant) | Marrow morphology “suspicious” for AML | Second BMT, azacitidine | CR (28 d) | +167 | 1.09 | 4 (1) | CCR | N/A | 812+ |
| 10 | 4 Years/F | AML MLL rearrangement | Haplo father RI | 271 post–2nd transplant (relapsed 180 d after 1st transplant) | BM blasts 0.08% (0.48% 3 mo later) | IFNa, DLI | MRD 0.01% (6 d) | +460 | 2.12 | 4 (1) | PR | 90 | 323 |
| 11 | 61 Years/M | MDS/AML EBB1, p53 mutation | MiniHaplo son NMA | 115 | BM blasts 20% | Azacitidine (post–TAA-T cells) | Persistent blasts (20 d) | +135 | 0.71 | 4 (1) | SD | 64 (PD) | 150 |
ACDVP16, cytarabine, daunorubicin, etoposide; B-ALL, B-cell acute lymphoblastic leukemia; BM, bone marrow; CNS, central nervous system; haplo, haploidentical; HIDAC, high-dose cytarabine; HLA = sibling, HLA-matched sibling; IT, intrathecal; MA, myeloablative; MDS, myelodysplastic syndrome; MTX, methotrexate; NMA, nonmyeloablative; Ph+, Philadelphia chromosome–positive; RI, reduced intensity; TBI, (total body irradiation; TKI, tyrosine kinase inhibitor; XRT, radiation therapy.
Twelve patients remaining in remission after BMT received TAA-T as preemptive therapy to prevent relapse for high-risk features including Philadelphia chromosome positivity (ALL), FLT3 positivity and mixed lineage leukemia (MLL) rearrangement (AML). Coadministration of chemotherapy and TKIs along with TAA-T was permitted throughout the study (Table 2). These 12 patients remained in CCR and were MRD− at the time of TAA-T infusion (Table 2).
Patient characteristics and disease response in patients treated preemptively with TAA-T infusion.
| Patient ID | Age/sex | Diagnosis and high-risk features | Donor and transplant type | Posttransplant treatment | Day post-BMT at time of TAA-T infusion | TAA-T dose level (number of doses) | ALC at first infusion (k/IU) | Best response postinfusion | Time to relapse postinfusion (days) | Survival postinfusion (days) |
|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 9 Years/F | AML MLL, NPM1 | HLA = sibling NMA | Azacitidine maintenance | +189 | 4 (1) | 0.52 | CCR | 134 | 728 |
| 13 | 31 Years/M | AML MLL t11:16 t7:11 der7 partial trisomy 11q ATM and WT1 mutation | HLA = sibling MA | None | +337 | 4 (1) | 1.82 | CCR | N/A | 806+ |
| 14 | 67 Years/F | AML/MDS | MiniHaplo NMA | None | +167 | 1 (1) | 3.27 | CCR | N/A | 561+ |
| 15 | 31 Years/M | AML NPM1, PTPN1 mutation | HLA = sibling NMA | None | +261 | 2 (1) | 0.98 | CCR | N/A | 368+ |
| 16 | 25 Years/M | AML FLT3 | HLA = sibling MA | Gilteritinib | +212 | 4 (1) | 1.30 | CCR | N/A | 383+ |
| 17 | 1 Years/F | AML MLL rearrangement | Haplo MA | None | +131 | 4 (1) | 4.09 | CCR | N/A | 771+ |
| 18 | 59 Years/M | AML monosomy 7 | Haplo MA | None | +92 | 4 (1) | 0.51 | CCR | 700 | 707+ |
| 19 | 47 Years/F | AML TP53 complex karyotype | Haplo NMA | None | +149 | 4 (1) | 0.64 | PD (MRD+ day 35) | 275 | 644 |
| 20 | 7 Years/F | AML inv(16) | HLA = sibling MA | None | +55 | 4 (1) | 1.30 | CCR | N/A | 380+ |
| 21 | 28 Years/F | AML leukemia cutis | Haplo NMA | None | +148 | 4 (1) | 2.20 | CCR | 64 | 228 |
| 22 | 18 Years/F | AML GATA2, germline mutation | Haplo NMA | None | +83 | 3 (1) | 1.10 | CCR | 126 | 126+ |
| 23 | 23 Years/M | AML relapsed pretransplant | HLA = sibling RI | None | +148 | 4 (1) | 0.57 | CCR | N/A | 189+ |
| Patient ID | Age/sex | Diagnosis and high-risk features | Donor and transplant type | Posttransplant treatment | Day post-BMT at time of TAA-T infusion | TAA-T dose level (number of doses) | ALC at first infusion (k/IU) | Best response postinfusion | Time to relapse postinfusion (days) | Survival postinfusion (days) |
|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 9 Years/F | AML MLL, NPM1 | HLA = sibling NMA | Azacitidine maintenance | +189 | 4 (1) | 0.52 | CCR | 134 | 728 |
| 13 | 31 Years/M | AML MLL t11:16 t7:11 der7 partial trisomy 11q ATM and WT1 mutation | HLA = sibling MA | None | +337 | 4 (1) | 1.82 | CCR | N/A | 806+ |
| 14 | 67 Years/F | AML/MDS | MiniHaplo NMA | None | +167 | 1 (1) | 3.27 | CCR | N/A | 561+ |
| 15 | 31 Years/M | AML NPM1, PTPN1 mutation | HLA = sibling NMA | None | +261 | 2 (1) | 0.98 | CCR | N/A | 368+ |
| 16 | 25 Years/M | AML FLT3 | HLA = sibling MA | Gilteritinib | +212 | 4 (1) | 1.30 | CCR | N/A | 383+ |
| 17 | 1 Years/F | AML MLL rearrangement | Haplo MA | None | +131 | 4 (1) | 4.09 | CCR | N/A | 771+ |
| 18 | 59 Years/M | AML monosomy 7 | Haplo MA | None | +92 | 4 (1) | 0.51 | CCR | 700 | 707+ |
| 19 | 47 Years/F | AML TP53 complex karyotype | Haplo NMA | None | +149 | 4 (1) | 0.64 | PD (MRD+ day 35) | 275 | 644 |
| 20 | 7 Years/F | AML inv(16) | HLA = sibling MA | None | +55 | 4 (1) | 1.30 | CCR | N/A | 380+ |
| 21 | 28 Years/F | AML leukemia cutis | Haplo NMA | None | +148 | 4 (1) | 2.20 | CCR | 64 | 228 |
| 22 | 18 Years/F | AML GATA2, germline mutation | Haplo NMA | None | +83 | 3 (1) | 1.10 | CCR | 126 | 126+ |
| 23 | 23 Years/M | AML relapsed pretransplant | HLA = sibling RI | None | +148 | 4 (1) | 0.57 | CCR | N/A | 189+ |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; Haplo, haploidentical donor; HLA = sibling, HLA-matched sibling; MA, myeloablative BMT; MDS, myelodysplastic syndrome; NMA, nonmyeloablative BMT; PD, progressive disease; RI, reduced intensity BMT.
Polyclonal effector and effector memory TAA-T recognizing WT1, PRAME, and survivin were expanded from healthy donors for clinical use
The median time from donor collection to product manufacture and release testing of the TAA-T products (n = 23) was 29 days (range, 20-41). The 23 patients treated received 1 to 4 infusions each, with a total of 29 infusions administered.
The phenotype of TAA-T products ultimately infused are shown in Figure 1A-C, with variable compositions of CD8+ T cells (median, 43.86%; range, 8.56% to 69.53%), CD4+ T cells (19.51%; 2.15% to 53.9%), and CD3−CD16+CD56+ cells (4.57%; 0.05% to 57.9%; Figure 1A). B cells and monocytes accounted for <2% (0.11%; 0% to 1.08%) of all final products (data not shown). Evaluable products (n = 11) were predominantly comprised of effector memory T-cell (CD3+CD4/8+CD45RO+CCR7−CD62L−) populations with a median of 68.75% of CD8+ (range, 13.08% to 87.97%) and 49.72% of CD4+ cells (range, 10.56% to 79.1%). Central memory T-cell (CD3+CD4/8+CD45RO+CCR7+CD62L+) populations, shown to be important for long-term persistence of adoptively transferred T cells in vivo,22 comprised a median of 3.8% of CD8+ (range, 1.13% to 19.23%) and 6.75% of CD4+ cells (range, 1.69% to 16.52%). CD8+ TEFF (CD3+CD8+CD45RO−CCR7−CD62L−) comprised a median of 2% (range, 0.2% to 8.6%), whereas minimal numbers (<1%) of CD4+ TEFF populations were detected in the infused products (Figure 1B-C). Finally, the TAA-T products showed broad TCR diversity, consistent with virus-specific T-cell products derived from naïve donors (Figure 1D).23-25
![Characterization of the TAA-T product by phenotype and TCR clonotype diversity. (A) Variable composition of the TAA-T products by phenotype (CD8+, CD4+, NK [natural killer], TCRγδ [γ δ T cells], NKT [natural killer T cells]) presented as percent of total cells in product as determined by 12-color flow cytometry (n = 23). (B-C) Memory phenotype described as central memory (TCM; CD3+CD4/8+CD45RO+CCR7+CD62L+), effector memory (TEM; CD3+CD4/8+CD45RO+CCR7−CD62L−), and effector T cells (TEFF; CD3+CD4/8+CD45RO−CCR7−CD62L) for evaluable samples (n = 11). (D) Diversity of TCR sequences of TAA-T products shown for representative patients in the relapsed group (P9, P3) and patients treated preemptively with TAA-T products (P12, P13).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/8/10.1182_bloodadvances.2021006831/3/m_advancesadv2021006831f1.png?Expires=1765883510&Signature=C7kvGRK~8NDVY9N7m71rdI-oecmUer6T1FiwwJnZt~dS9F8YPWxnGgQLfPpEK-mU0zXBe4K96UZupnbfnv-rASfq1p76ikhOIJBNEH7XN03FoGE0GTj-lFv3geQr9xnTeqcF0nI0Murf8KqYQaqHmq8OVsraH56JhJh-QxxyfxSLcQUlHYicHeE8us6Y6cN4eF8Imddj5R-EQAeytwflWqFdYv9cKs-5yXnh~uW8cDM1S~S82T3~4k~XayD1N0uwVa4yUwB3JIcCEObfA-7a7nzkuLQThkLxxA6dD664-rjgsOKKhCJjpWo5xACD4RavW82NEFj6lQ2FSTujCWbonA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of the TAA-T product by phenotype and TCR clonotype diversity. (A) Variable composition of the TAA-T products by phenotype (CD8+, CD4+, NK [natural killer], TCRγδ [γ δ T cells], NKT [natural killer T cells]) presented as percent of total cells in product as determined by 12-color flow cytometry (n = 23). (B-C) Memory phenotype described as central memory (TCM; CD3+CD4/8+CD45RO+CCR7+CD62L+), effector memory (TEM; CD3+CD4/8+CD45RO+CCR7−CD62L−), and effector T cells (TEFF; CD3+CD4/8+CD45RO−CCR7−CD62L) for evaluable samples (n = 11). (D) Diversity of TCR sequences of TAA-T products shown for representative patients in the relapsed group (P9, P3) and patients treated preemptively with TAA-T products (P12, P13).
Characterization of the TAA-T product by phenotype and TCR clonotype diversity. (A) Variable composition of the TAA-T products by phenotype (CD8+, CD4+, NK [natural killer], TCRγδ [γ δ T cells], NKT [natural killer T cells]) presented as percent of total cells in product as determined by 12-color flow cytometry (n = 23). (B-C) Memory phenotype described as central memory (TCM; CD3+CD4/8+CD45RO+CCR7+CD62L+), effector memory (TEM; CD3+CD4/8+CD45RO+CCR7−CD62L−), and effector T cells (TEFF; CD3+CD4/8+CD45RO−CCR7−CD62L) for evaluable samples (n = 11). (D) Diversity of TCR sequences of TAA-T products shown for representative patients in the relapsed group (P9, P3) and patients treated preemptively with TAA-T products (P12, P13).
TAA-T were antigen specific, polyfunctional, and cytolytic against leukemia blasts in vitro
Antigen specificity of all TAA-T products (n = 23) was determined by anti-IFNγ ELISpot assay prior to initial infusion and was repeated on evaluable products prior to subsequent infusions (n = 25; Figure 2A). All but 1 product demonstrated response to the SEB+ control. There was insufficient sample to repeat the analysis to evaluate for failure of the assay. Median (and range) antigen-specific responses (IFNγ production per 1 × 105 cells) were evaluated for WT1 (16 spot-forming units [SFU]; 0-450 SFU), PRAME (39 SFU; 0-523), survivin (10 SFU; 0-114), and all 3 peptides combined (TAA; 44 SFU; 0-582). The median negative control (actin) was 12 SFU (range: 0-144) and the median positive control (SEB) was 732 SFU (range, 1 to too many to count). Mean antigen responses (IFNγ production per 1 × 105 cells) were statistically significantly different from actin (negative control) for WT1 (P = .0469), PRAME (P = .0001), and TAA (P < .0001) but not for survivin (P = .7028). A positive antigen response in each product was classified as an IFNγ production greater than the upper 95% confidence interval (CI) of the median IFNγ production to actin, the negative control. These results similarly identify PRAME as the target antigen with the most consistently positive response across products. This data is shown in supplemental Results.
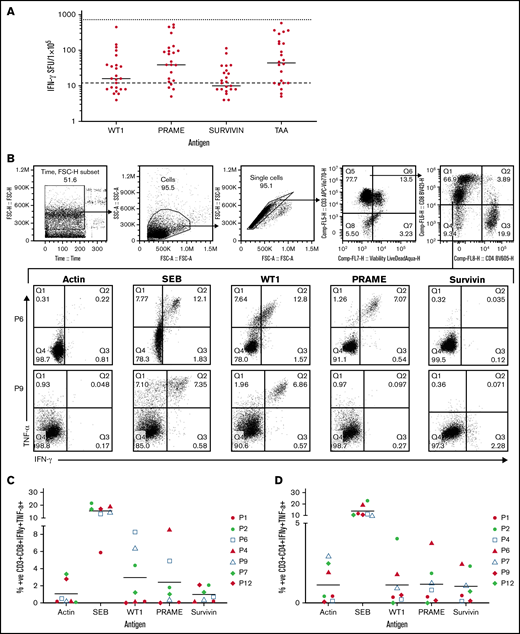
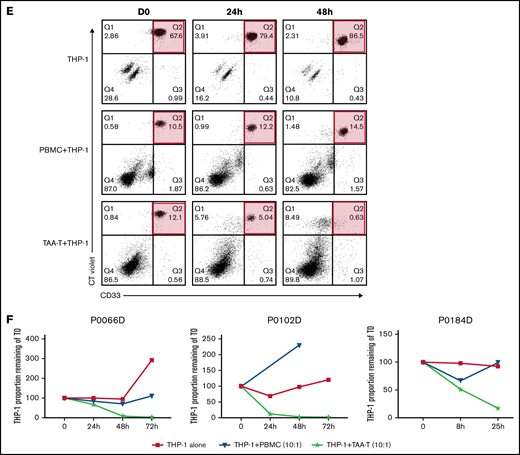
Antigen specificity as measured by anti-IFNγ ELISpot, TNFα and IFNγ intracellular cytokine staining, and cytolytic function of the TAA-T products. (A) Target antigen specificity of the TAA-T product (n = 25) as determined by IFNγ production, measured by ELISpot. Target antigens were WT1, PRAME, survivin, and TAA (WT1, PRAME, and survivin pepmixes combined). The bottom dotted line denotes the median for negative control (actin = 12 SFU), the top dotted line denotes the median for positive control (SEB = 732 SFU). Mean antigen responses were statistically significantly different from actin for WT1 (P = .0469), PRAME (P = .0001), and TAA (P < .0001) but not for survivin (P = .7028). (B) TNFα and IFNγ intracellular cytokine staining (ICS) demonstrates antigen specificity for WT1 and PRAME shown for products (P6, P9). Antigen specificity measured by TNFα and IFNγ ICS of CD8+ T cells (C) and CD4+ T cells (D) of the TAA-T-cell product in evaluable samples (n = 7). SEB is used as the positive control and actin as negative control. (E) In vitro cytolytic activity of the HLA A*02+ TAA-T product against an HLA A*02+ AML cell line (THP-1) as compared with a donor lymphocyte infusion (donor PBMCs) product. (F) Superior cytolytic activity against THP-1 Violet+ CD33+ cells of the TAA-T product as compared with donor lymphocyte infusion (PBMC) is reproducible in the majority of A*02+ donor TAA-T products evaluated (as shown for P2, P6, P12). SEB, staphylococcal enterotoxin B.
Antigen specificity as measured by anti-IFNγ ELISpot, TNFα and IFNγ intracellular cytokine staining, and cytolytic function of the TAA-T products. (A) Target antigen specificity of the TAA-T product (n = 25) as determined by IFNγ production, measured by ELISpot. Target antigens were WT1, PRAME, survivin, and TAA (WT1, PRAME, and survivin pepmixes combined). The bottom dotted line denotes the median for negative control (actin = 12 SFU), the top dotted line denotes the median for positive control (SEB = 732 SFU). Mean antigen responses were statistically significantly different from actin for WT1 (P = .0469), PRAME (P = .0001), and TAA (P < .0001) but not for survivin (P = .7028). (B) TNFα and IFNγ intracellular cytokine staining (ICS) demonstrates antigen specificity for WT1 and PRAME shown for products (P6, P9). Antigen specificity measured by TNFα and IFNγ ICS of CD8+ T cells (C) and CD4+ T cells (D) of the TAA-T-cell product in evaluable samples (n = 7). SEB is used as the positive control and actin as negative control. (E) In vitro cytolytic activity of the HLA A*02+ TAA-T product against an HLA A*02+ AML cell line (THP-1) as compared with a donor lymphocyte infusion (donor PBMCs) product. (F) Superior cytolytic activity against THP-1 Violet+ CD33+ cells of the TAA-T product as compared with donor lymphocyte infusion (PBMC) is reproducible in the majority of A*02+ donor TAA-T products evaluated (as shown for P2, P6, P12). SEB, staphylococcal enterotoxin B.
Antigen specificity was also evaluated using ICS for TNFα and IFNγ by flow cytometry for evaluable products (n = 7; Figure 2B-D; supplemental Results).
Finally, the specific cytolytic function of products generated for HLA-A*02+ patients with AML was evaluated using a coculture assay against the HLA-A*02+ AML cell line THP-1. The in vitro cytolytic activity of the TAA-T product against Violet+ CD33+ THP-1 cells was superior to that of the pseudo DLI product (PBMCs; Figure 2E). These results were reproduced in 3 other A*02+ donor-derived TAA-T products evaluated (Figure 2F).
TAA-T administration following allogeneic BMT was safe
After confirming eligibility, TAA-T were infused as outpatients, with no immediate infusion-related toxicities or adverse events. One patient withdrew from the study on day 23 postinfusion with bacterial sepsis and died of complications unrelated to TAA-T, leaving the recipients of 22 products evaluable for toxicity monitoring over a 45-day period (Tables 1 and 2; supplemental Table 1). No patients developed cytokine release syndrome or neurotoxicity attributable to TAA-T. Infection and alterations in renal function occurring during the 45-day safety monitoring period were not deemed attributable to TAA-T. Two patients developed elevated c-reactive protein, and 1 developed leukopenia postinfusion, likely related to TAA-T. Two patients developed dose-limiting liver toxicity with alterations in hepatic enzymes and bilirubin that were possibly attributable to TAA-T infusion and fully resolved with treatment.
Ten patients were diagnosed with acute and/or chronic GVHD posttransplant prior to TAA-T. However, only 4 patients developed GVHD of any grade after TAA-T infusion, with only 1 patient (P9) developing severe (grade 3) GVHD (stage 3 liver, stage 2 skin), which was diagnosed 4 weeks post–TAA-T (4.0 × 107/m2; dose level 4) and was associated with a concurrent Haemophilus influenzae B and Varicella zoster infection. This patient’s liver biopsy was indeterminant for the underlying cause of the liver toxicity, favoring drug injury vs GVHD. However, this was characterized as a dose-limiting, grade 4 liver toxicity possibly related to the TAA-T. Systemic steroids, tacrolimus, and extracorporeal photopheresis in addition to antiviral treatment successfully controlled the liver toxicity, and he remains in remission >1 year post–TAA-T. Three other patients developed grades 1 and 2 skin GVHD post–TAA-T (at dose levels 1, 3, and 4); all were steroid responsive. One patient with grade 2 skin GVHD developed a dose-limiting toxicity with grade 4 transaminitis possibly attributed to the TAA-T.
Disease response post–TAA-T infusion (all patients)
Seventeen of 23 patients (73.9%) with acute leukemia were in continued complete hematologic (morphologic) remission in the first 45 days following TAA-T. The median duration of survival postinfusion for all patients was 644 days at the time of submission. The 1-year OS for all patients with AML (n = 20) was 66.4% (95% CI, 39.84-83.33) post–TAA-T.
Disease outcomes for patients who relapsed after first BMT
Nine of 11 patients who relapsed post-BMT were in complete morphologic remission at the time of TAA-T infusion. One patient had detectable disease after achieving a PR in response to bridging chemotherapy, and 1 patient had disease progression at the time of TAA-T. The 1-year OS after TAA-T for patients who relapsed after transplant (n = 11) was 36.4% (95% CI, 11.2-62.7). Median survival was 255 days (including P1, who died of sepsis and was not evaluable for response at week 6) with 3 long-term survivors (B-ALL [n = 1], AML [n = 2]) at 812 to 1160 days postinfusion (Figure 3A). LFS 1 year post–TAA-T infusion was 27.3%, and median LFS was 64 days.
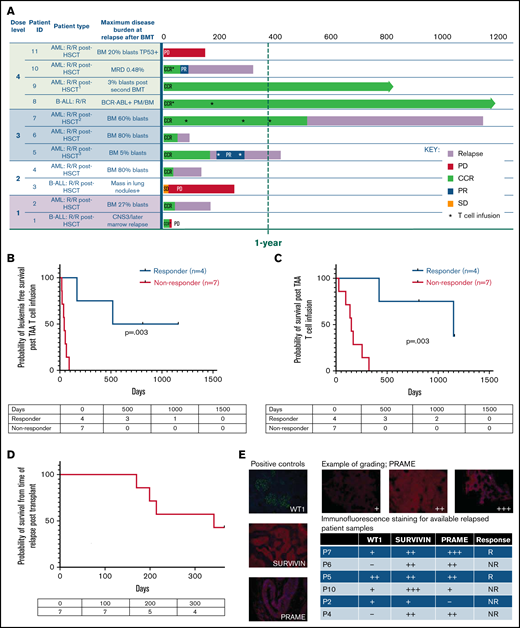
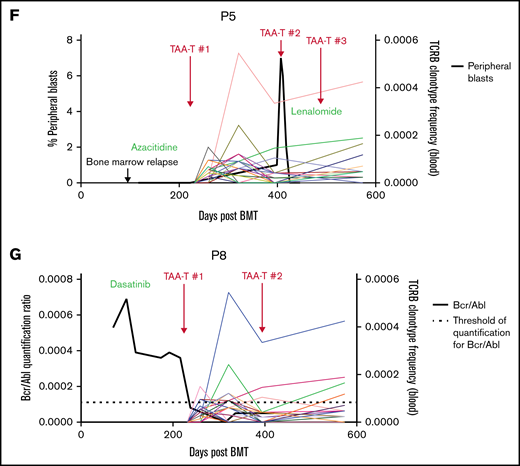
Clinical outcomes for patients treated with TAA-T for relapsed disease after BMT (n = 11). (A) Swimmer plot showing clinical outcomes following salvage therapy and TAA-T infusion in patients with relapsed/refractory disease after BMT, categorized by dose level (1-4). Hematologic remission was achieved in 9/11 patients prior to TAA-T infusion with postinfusion clinical outcomes defined as CCR, PR, stable disease (SD), PD, and relapse. Patients in hematologic remission with MRD are noted as CCR*. Patients who did not achieve hematologic remission are noted as + (P3, P11). The dotted line denotes 1 year postinfusion. (B) Kaplan-Meier curve estimating LFS postinfusion of relapsed patients. One-year LFS 27.3%; median LFS was 64 days. Patients characterized as responders (CCR within 3 months of first TAA-T infusion; n = 4) had prolonged median LFS (839 days) compared with nonresponders (PD/R within 3 months of first TAA-T infusion; n = 7); median LFS was 42 days (P = .003). (C) Kaplan-Meier curve estimating OS postinfusion of relapsed patients. One-year OS was 36.36% with median survival of 255 days post–TAA-T infusion. Responders had prolonged median OS (1150 days) compared with nonresponders (150 days) (P = .003). (D) One-year postrelapse OS was 42% in early relapsers (patients with relapse within 6 months of transplant; n = 7) who received TAA-T infusion. (E) Qualitative grading of immunofluorescence expression of TAA targets (WT1, PRAME, and survivin) on blast population and clinical outcomes following TAA-T of evaluable patients with relapsed AML posttransplant. The paraffin-embedded tissues were deparaffinized and incubated post–antigen retrieval with anti-survivin, anti–Wilms tumor protein (abcam), and anti-PRAME (Sigma) followed by Alexa Fluor568 (Texas red channel) donkey anti-rabbit IgG secondary antibody for survivin and PRAME (abcam) and AlexaFluor488 (FITC) donkey anti-mouse IgG secondary antibody for WT1 (abcam). The sections were mounted with DAPI staining solution (abcam), and the images were captured at 20× magnification on an Olympus BX53-DP73 microscope using cellSens software. Clinical outcomes characterized as responder and nonresponder (as above). (F) Disease course and TCR unique clonotype frequencies over time for P5 with MDS/AML, relapsed 117 days posttransplant and subsequently achieved CR with salvage therapy (azacitidine) prior to TAA-T infusion. Hematologic relapse with peripheral blasts cleared with a second TAA-T infusion, azacitidine, and lenalidomide, though remained MRD+. (G) Disease course and unique TCR clonotype frequency over time for P8, a pediatric patient with Ph+ B-cell ALL with persistent BCR/ABL positivity posttransplant despite treatment with dasatinib. Briefly achieved BCR/ABL negativity following first TAA-T infusion followed by rise in BCR/ABL quantification ratio following the second TAA-T infusion. DAPI, 4′,6-diamidino-2-phenylindole; IgG, immunoglobulin G.
Clinical outcomes for patients treated with TAA-T for relapsed disease after BMT (n = 11). (A) Swimmer plot showing clinical outcomes following salvage therapy and TAA-T infusion in patients with relapsed/refractory disease after BMT, categorized by dose level (1-4). Hematologic remission was achieved in 9/11 patients prior to TAA-T infusion with postinfusion clinical outcomes defined as CCR, PR, stable disease (SD), PD, and relapse. Patients in hematologic remission with MRD are noted as CCR*. Patients who did not achieve hematologic remission are noted as + (P3, P11). The dotted line denotes 1 year postinfusion. (B) Kaplan-Meier curve estimating LFS postinfusion of relapsed patients. One-year LFS 27.3%; median LFS was 64 days. Patients characterized as responders (CCR within 3 months of first TAA-T infusion; n = 4) had prolonged median LFS (839 days) compared with nonresponders (PD/R within 3 months of first TAA-T infusion; n = 7); median LFS was 42 days (P = .003). (C) Kaplan-Meier curve estimating OS postinfusion of relapsed patients. One-year OS was 36.36% with median survival of 255 days post–TAA-T infusion. Responders had prolonged median OS (1150 days) compared with nonresponders (150 days) (P = .003). (D) One-year postrelapse OS was 42% in early relapsers (patients with relapse within 6 months of transplant; n = 7) who received TAA-T infusion. (E) Qualitative grading of immunofluorescence expression of TAA targets (WT1, PRAME, and survivin) on blast population and clinical outcomes following TAA-T of evaluable patients with relapsed AML posttransplant. The paraffin-embedded tissues were deparaffinized and incubated post–antigen retrieval with anti-survivin, anti–Wilms tumor protein (abcam), and anti-PRAME (Sigma) followed by Alexa Fluor568 (Texas red channel) donkey anti-rabbit IgG secondary antibody for survivin and PRAME (abcam) and AlexaFluor488 (FITC) donkey anti-mouse IgG secondary antibody for WT1 (abcam). The sections were mounted with DAPI staining solution (abcam), and the images were captured at 20× magnification on an Olympus BX53-DP73 microscope using cellSens software. Clinical outcomes characterized as responder and nonresponder (as above). (F) Disease course and TCR unique clonotype frequencies over time for P5 with MDS/AML, relapsed 117 days posttransplant and subsequently achieved CR with salvage therapy (azacitidine) prior to TAA-T infusion. Hematologic relapse with peripheral blasts cleared with a second TAA-T infusion, azacitidine, and lenalidomide, though remained MRD+. (G) Disease course and unique TCR clonotype frequency over time for P8, a pediatric patient with Ph+ B-cell ALL with persistent BCR/ABL positivity posttransplant despite treatment with dasatinib. Briefly achieved BCR/ABL negativity following first TAA-T infusion followed by rise in BCR/ABL quantification ratio following the second TAA-T infusion. DAPI, 4′,6-diamidino-2-phenylindole; IgG, immunoglobulin G.
Patients characterized as “responders” (defined as CCR 3 months after TAA-T) had a median LFS of 839 days vs 42 days in the “nonresponder” (defined as relapse or PD in the first 3 months after TAA-T) group (P = .003; Figure 3B). The median OS in the responders was 1150 days, compared with 150 days in the nonresponder group (P = .003; Figure 3C). Notably, in the poorest prognosis group (patients with hematologic or extramedullary relapse ≤6 months post-BMT who did not undergo a second transplant prior to TAA-T [n = 7]), the median survival was 159 days with a 1-year OS of 25% after infusion (95% CI, 3.7-55.8) and 1-year OS of 42.8% from time of relapse (Figure 3A-D).
Immunofluorescence staining to evaluate expression of targeted antigens (WT1, PRAME, and survivin) on the blast populations from evaluable patients showed varying degrees of positivity. No inferences could be made with respect to the correlation of immunofluorescent antigen positivity and disease response because of the small sample size (Figure 3E; supplemental Figure 5). Individual courses of representative patients who had relapsed after first BMT and had demonstrable responses with in vivo T-cell expansion after TAA-T are shown in Figure 3F-G.
Disease outcomes for patients at high risk for relapse post-BMT
The median survival in patients defined as high risk for relapse and treated with TAA-T as preemptive therapy (n = 12) was not yet reached at the time of submission, with 9/12 patients remaining alive. Eight of the 9 (88.9%) evaluable patients were alive at 1 year, and of those, 5 (62.5%) remained alive and in remission at the time of submission (Figure 4A). Nine patients were in persistent remission (no relapse within 6 months of TAA-T), with 2 patients relapsing at 275 and 700 days, respectively. Patient P19, who relapsed at 275 days, subsequently underwent a second transplant from a second donor and ultimately died of disease progression 644 days postinitial TAA-T (Figure 4A). Three patients with AML relapsed within 6 months of TAA-T (Figure 4B), and 2 of these patients died at 228 and 728 days, respectively, postinfusion. The third patient remains alive 126 days post–TAA-T (Figure 4C). None of the patients treated preemptively had “early” relapse (defined as relapse within 6 months after first transplant). This cohort experienced a median LFS of 792 days and median OS of 917 days after first transplant (supplemental Figure 7A-B).
![Clinical outcomes for patients with high-risk disease treated preemptively with TAA-T after BMT (n = 12). (A) Swimmer plot showing clinical outcomes of patients treated preemptively with TAA-T infusion for high-risk disease after BMT, categorized by dose level (1-4). All patients were in CCR at the time of TAA-T infusion. The dotted line denotes 1 year postinfusion. (B) Kaplan-Meier curve estimating LFS postinfusion of preemptively treated patients. Median LFS has not been reached for all patients. Patients who relapsed in the first 6 months post–TAA-T infusion (n = 2) had median LFS of 99 days; median LFS for patients in persistent remission (no relapse or PD within 6 months of TAA-T infusion [n = 9]) has not been reached. (C) Kaplan-Meier curve estimating OS postinfusion of preemptively treated patients. Median OS has not been reached for all patients.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/8/10.1182_bloodadvances.2021006831/3/m_advancesadv2021006831f4.png?Expires=1765883510&Signature=FZOfQSqCm4XMoD72Wm4dSuz15~zV5WA8SUqEBrxEWussv2mE3OwKM47bFtfNQbUcp-qspczEnhAF2egkG9l9--Msaq9A4XOFJnvvH1f5Bs5aies4P-hLnLBq~Bu~mLJwzyaayNz1DiA7gA53BlpVAKOZtZ6gfBj4Dl4Wn2ZPcY4Q3Dunww~ewfFgoKa5ivf0WrCyabikTcrYszMbVR5qW3IaowopAikMe4G0OfFAkDb9JSLf6e2ViEhAhp6e0ljZwdL7gjSEC6x6SovAhPB~rCGrEol-3dObYVaAC3JaKUuz84Ap4zG2s3VRmmcZTrlOcIFsC57gXPhUiGV2gyjzag__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clinical outcomes for patients with high-risk disease treated preemptively with TAA-T after BMT (n = 12). (A) Swimmer plot showing clinical outcomes of patients treated preemptively with TAA-T infusion for high-risk disease after BMT, categorized by dose level (1-4). All patients were in CCR at the time of TAA-T infusion. The dotted line denotes 1 year postinfusion. (B) Kaplan-Meier curve estimating LFS postinfusion of preemptively treated patients. Median LFS has not been reached for all patients. Patients who relapsed in the first 6 months post–TAA-T infusion (n = 2) had median LFS of 99 days; median LFS for patients in persistent remission (no relapse or PD within 6 months of TAA-T infusion [n = 9]) has not been reached. (C) Kaplan-Meier curve estimating OS postinfusion of preemptively treated patients. Median OS has not been reached for all patients.
Clinical outcomes for patients with high-risk disease treated preemptively with TAA-T after BMT (n = 12). (A) Swimmer plot showing clinical outcomes of patients treated preemptively with TAA-T infusion for high-risk disease after BMT, categorized by dose level (1-4). All patients were in CCR at the time of TAA-T infusion. The dotted line denotes 1 year postinfusion. (B) Kaplan-Meier curve estimating LFS postinfusion of preemptively treated patients. Median LFS has not been reached for all patients. Patients who relapsed in the first 6 months post–TAA-T infusion (n = 2) had median LFS of 99 days; median LFS for patients in persistent remission (no relapse or PD within 6 months of TAA-T infusion [n = 9]) has not been reached. (C) Kaplan-Meier curve estimating OS postinfusion of preemptively treated patients. Median OS has not been reached for all patients.
TCR clonotype expansion and persistence
TCR sequencing was performed on available TAA-T products (n = 11) and on recipient peripheral blood samples obtained preinfusion vs postinfusion (range, 28-365 days postinfusion). Evaluation of unique TCR clonotypes (present in the product but not the recipient prior to infusion) at postinfusion timepoints demonstrated expansion of unique TCR clonotypes in patients receiving TAA-T after relapse (Figure 3F-G; supplemental Figure 6A-D) and for high-risk disease (supplemental Figure 6E-F). The unique T-cell clones were shown to persist up to 6 months postinfusion in the relapsed group and up to 1 year in the preemptively treated group.
Discussion
This study found that administration of donor-derived TAA-T targeting WT1, PRAME, and survivin was safe and feasible for patients with high-risk leukemia after allogeneic BMT. Eleven patients had relapsed prior to TAA-T infusion, 9 of whom relapsed within 6 months of transplant or had residual/refractory disease. The addition of TAA-T to treat or preempt posttransplant relapse in this subset of patients was accompanied by few treatment-related serious adverse events and a 1-year OS of 36% and LFS of 27.3% in the relapsed group. Moreover, in patients who relapsed <6 months post-BMT and subsequently received TAA-T, their 1-year OS from time of relapse after first transplant was 42.8%. Treatment failure and death is due in part to an insufficient GVL effect, dysregulation of immune fuction,26 and from toxicity of chemotherapy. McIver et al found that patients who relapse <6 months after allogeneic BMT had a <5% probability of survival irrespective of whether second BMT or less intensive management was chosen,3 with a 0% OS at 1 year after relapse in the patients who underwent a second transplant. Patients who had late relapse (>6 months post–first transplant) and subsequently underwent a second transplant, DLI with or without chemotherapy or chemotherapy alone had a 1-year OS of 33% after relapse,3 similar to other studies also showing later relapse associated with improved outcomes.5,6,10,27 Our cohort of patients with “early” relapse (defined as relapsing within 6 months of transplant) who did not undergo a second transplant prior to TAA-T infusion fared better, with less relapse and appreciably longer survival: 1-year OS from time of relapse after first transplant was 42.8% (Figure 3D) compared with 0%. These outcomes were more comparable to those with late relapse in the McIver cohort, although in the time since that publication, development of other novel therapies used the in the management of this cohort likely also contributed to the improved outcomes observed. Hence, these results are comparable or superior to similar cohorts of AML and ALL patients treated with alternative therapies,4 -6,9,10,28,29 suggesting that donor-derived TAA-T are at least as effective as chemotherapy and DLI, or second transplant, for the treatment of post-BMT relapsed leukemia but with appreciably less toxicity, particularly with respect to GVHD.5,6,10,27
Donor-derived TAA-T infusions provide an opportunity to expand a subset of cytotoxic T cells specific to tumor antigen targets while greatly reducing the risk of GVHD as compared with DLI.1,7,30 Extensive clinical experience with T cells expanded ex vivo with peptide antigens specific for the treatment of viral infections post-BMT indicate that such T cells lack alloreactivity and seldom, if ever, cause GVHD. The safety profile of the TAA-T product described herein is in line with these observations and also contrasts with the frequent occurrence of cytokine release syndrome and neurotoxicity, which complicates CD19 CAR-T used for relapsed ALL.
A recent study evaluated the effect of a different tumor-associated, antigen-specific T-cell product for patients with AML refractory to salvage chemotherapy with active disease at the time of infusion.31 Two of 6 patients had PR or CR to cell therapy alone, with 3 patients treated with demethylating agents and T cells in the adjuvant setting.31 In contrast, the majority of the relapsed patients in our study achieved CR with salvage chemotherapy prior to TAA-T. Several (n = 4) were treated with decitabine or azacitadine as bridging therapy, which has been shown to increase expression of some TAAs, including PRAME,32,33 and potentiate in vitro killing of AML blasts by PRAME-specific T cells.32 Therefore, further studies comparing patients receiving TAA-T therapy in combination with hypomethylating agents to those receiving TAA-T infusions with alternative or no salvage therapy are warranted to assess any synergistic effect these agents may have.
Key issues in designing and expanding effective antileukemic T cells is the selection of appropriate TAAs ubiquitously present on malignant cells and, ideally, not expressed on healthy human tissues. We chose to expand T cells against multiple TAAs (WT1, PRAME, and survivin) to target most malignancies through at least 1 expressed antigen. Of the available samples, the recipient’s malignancy expressed at least 1 of these TAAs by immunofluorescence (Figure 3E). However, preliminary data from this phase 1 study is not sufficient to analyze a potential correlation between immunofluorescent antigen positivity and disease response to TAA-T. Antigen specificity of the TAA-T products as evaluated by ELISpot demonstrated robust positivity for PRAME and TAA but not consistently for WT1 and survivin across all products. Similarly, CD8+ T-cell responses evaluated by ICS were variable across all patient products, with WT1 and PRAME trending above that of the negative control, although not statistically significant. This represents an opportunity for increasing the potency of the product for future studies by ensuring equal targeting of all antigens in this single product. Targeting multiple TAAs reduces the risk of tumor escape through downregulation of one antigen, but restricting to a maximum of TAAs also reduces the risk of antigenic competition observed with increased antigen targeting.34 Response to TAA-T infusions may also correlate directly with the number of target antigens expressed on the recipients’ tumor cells, with the poorest responses anticipated in patients not expressing any of the 3 TAA targets. This hypothesis remains to be definitively tested in larger, later-phase trials.
Growing experience with T-cell infusions to treat malignancy indicates that antitumor efficacy is enhanced by lymphodepletion and by persistence of the infused cells.35,36 We did not deliberately use prescribed lymphodepletion before TAA-T infusion, but the therapeutic effect of our TAA-T product may have been enhanced by lymphopenia from “bridging” chemotherapy or prior transplantation. Moreover, characterization of TCR sequencing postinfusion may predict benefit of TAA-T. Patients with disease response following TAA-T infusion had evidence of both persistence and expansion of TCR clonotypes that were present in the product but not present prior to infusion. However, given the small sample size and correlative nature of the data, we are not able to make any conclusions of causality. In future studies, identification and tracking of target antigen-specific unique clonotypes from the T-cell product and demonstration of cytotoxicity of the product against individual patient samples could provide further support of this hypothesis.
Finally, 2 patients were treated concomitantly with the TKI dasatinib for Philadelphia chromosome–positive ALL. This therapy was continued before and after TAA-T infusion and therefore may have suppressed antigen expression and activation of the T-cell product.37,38 One of these patients had refractory disease and died early in the follow-up time period. The second patient had prolonged survival and remains alive at the time of submission.
Rapid progress has been made in the manufacturing of highly effective leukemia-specific T cells. Notably, CD19 CAR-T have achieved spectacular results in B-cell malignancies with apparent cures of ALL relapsing post-BMT.11,12 However, they have appreciable postinfusion toxicity, and tumor escape can occur from downregulation of CD19.39,40 In the absence of suitable myeloid surface antigens, CAR-T therapy for AML remains more challenging and still under early stages of investigation. Our non–genetically engineered TAA-T product is therefore of particular relevance for patients with high-risk AML.
In conclusion, this phase 1 study demonstrates the safety and tolerability of donor-derived TAA-T-cell infusions following allogeneic BMT for the treatment of patients with high-risk/relapsed/refractory hematologic malignancies. Further studies are needed to determine long-term clinical disease outcomes and to optimize the dose and timing of TAA-T infusions posttransplant, particularly for preemptively treated patients. However, these data provide robust evidence that donor-derived TAA-T infusion is a well-tolerated therapy. Later-phase studies are warranted to further evaluate the effect of TAA-T on prolonging remission in patients with high-risk hematologic malignancies post-BMT.
Acknowledgments
The authors would like to acknowledge the work of Maria Martin Manso for her role in T-cell product manufacturing and Sean Gillen for study coordination in the early stages of this trial.
This work was supported by National Cancer Institute (NCI), National Institutes of Health (NIH), grant P01-CA-015396 (R.J.J., C.M.B., and K.R.C.), a Leukemia and Lymphoma Society SCOR 7018-04 (C.M.B., K.M.W. and P.J.H.), a Hyundai Hope on Wheels grant (C.M.B. and K.M.W.), Ben’s Run Foundation (C.M.B. and K.M.W.), Rising Tides Foundation (K.M.W. and C.M.B.), and a Ruth L. Kirschstein National Research Service Award Institutional Research Training Grant awarded to the Children’s Research Institute Hematology Training Program by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (5T32HL110841-08) (H.K.). This project was supported by a grant from the NIH National Center for Advancing Translational Sciences (UL1TR001876).
Authorship
Contribution: This paper was primarily written by H.K., K.R.C., A.J.B., R.J.J., and C.M.B. The study was developed and designed by R.J.J., K.M.W., and C.M.B. The CNH principal investigators (PIs) on the clinical trial were J.S.D., D.J., and K.M.W., and the Johns Hopkins University site PI was K.R.C. The Investigational New Drug Application (IND) was held by C.M.B. Programmatic oversight and quality assurance was provided by F.H., M.F., K.F., D.K. D.J., J.S.D., K.R.C., K.M.W., A.J.B., R.J.J. cared for the transplantation patients enrolled on the trial and were responsible for data integrity and capture. Cells were generated and evaluated by P.J.H., A.S., A.D., J.T., and N.Z. Regulatory oversight was performed by F.H. Data analysis was accomplished by H.K., M.G., C.R.C., H. Lang, M.S., and K.M.W. Statistical studies were performed by H. Liang, A.Z., and H.K. All authors reviewed the manuscript and made the decision to submit for publication, vouching for the accuracy and completeness of the data reported and fidelity to the protocol.
Conflict-of-interest disclosure: C.M.B. has stock or ownership in Cabaletta Bio, Catamaran Bio, and Neximmune. C.M.B. also has equity interest in Mana Therapeutics, which subsequently licensed the technology used in this study. As sponsor of the study, she helped design the clinical trial but was not responsible for final decision-making regarding data collection, interpretation of outcome data, or decision to publish. P.J.H. and C.R.C. also have stock or ownership in Mana Therapeutics and serve on the board of directors (P.J.H.) or scientific advisory board (C.R.C.) and were not responsible for clinical trial design, data collection, interpretation of outcome data, or decision to publish. P.J.H. is also on the scientific advisory board of Cellevolve and an advisor for Maxcyte. The remaining authors declare no competing financial interests.
This study is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. The funding sources had no role in trial design, data collection, interpretation of data, or decision to publish.
Correspondence: Catherine M. Bollard, Children’s National Health System, The George Washington University, 111 Michigan Ave, NW, Washington, DC 20010; e-mail: cbollard@childrensnational.org; and Richard J. Jones, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Johns Hopkins University School of Medicine, 401 N Broadway, Baltimore, MD 21231; e-mail: rjjones@jhmi.edu.

