

Abstract
Anemia after allogeneic hematopoietic stem cell transplantation (HSCT) can be immune or non–immune mediated. Auto- or alloimmunity resulting from blood group incompatibility remains an important cause in post-HSCT immune-mediated anemia. ABO incompatibility is commonly encountered in HSCT and may lead to serious clinical complications, including acute hemolysis, pure red cell aplasia, and passenger lymphocyte syndrome. It remains controversial whether ABO incompatibility may affect HSCT outcomes, such as relapse, nonrelapse mortality, graft-versus-host disease, and survival. Non-ABO incompatibility is less frequently encountered but can have similar complications to ABO incompatibility, causing adverse clinical outcomes. It is crucial to identify the driving etiology of post-HSCT anemia in order to prevent and treat this condition. This requires a comprehensive understanding of the mechanism of anemia in blood group–incompatible HSCT and the temporal association between HSCT and anemia. In this review, we summarize the literature on post-HSCT immune-mediated anemia with a focus on ABO and non-ABO blood group incompatibility, describe the underlying mechanism of anemia, and outline preventive and treatment approaches.
Introduction
There are at least 43 recognized blood groups in humans involving 345 red blood cell (RBC) antigens.1 Approximately 30% to 50% of hematopoietic stem cell transplantations (HSCTs) are performed across the ABO blood group barrier, and clinically significant hemolysis is encountered in 10% to 15% of cases.2,3 Other complications of ABO-mismatched HSCT include hemolytic infusion reactions, delayed engraftment, and pure red cell aplasia (PRCA).4,5 Compared with those of ABO-mismatched HSCT, complications of non-ABO blood group–mismatched HSCT are less well-characterized. Recipients and donors are not routinely checked for non-ABO blood group phenotypes unless clinically indicated, such as when there is a known history of alloantibodies or heavy transfusion requirement in the recipient before transplantation.6 The incidence of non-ABO blood group mismatch–related immunohematologic complications after HSCT is reported to be <10%, which likely reflects underreporting.7-10
Auto- or alloimmunity against RBCs is an important etiology of immune-mediated anemia in blood group–mismatched HSCT. The source of auto- or alloimmunity may be donor or recipient related, including passive antibody transfusion, passenger lymphocyte syndrome (PLS), alloantibodies formed against engrafting hematopoietic cells, or new auto- or alloantibodies resulting from transfusions (Figures 1 and 2). Antibodies can mediate phagocytosis and cellular toxicity and activate the complement pathway, resulting in intra- and extravascular hemolysis.11 Immune-mediated cytopenia can also occur as a manifestation of chronic graft-versus-host disease (GVHD).12,13 The underlying mechanism of cytopenia in GVHD is unclear, but it is likely to result from altered T-cell immune reconstitution.14
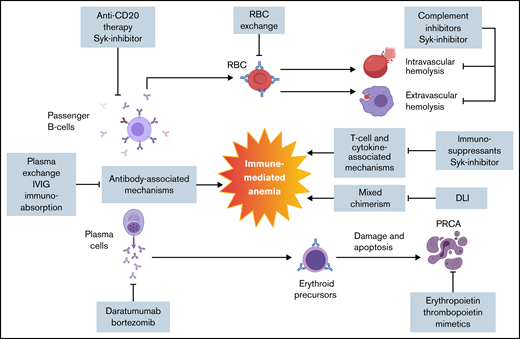
Pathophysiology and targeted therapy for post-HSCT immune-mediated anemia. Post-HSCT anemia is multifactorial as a result of new auto- or alloantibodies in combination with T cell– and cytokine-mediated inflammatory processes. There are no formal guidelines in management. Several therapeutic approaches that target different pathophysiologic aspects of post-HSCT immune-mediated anemia are outlined. DLI, donor lymphocyte infusion; IVIG, IV immunoglobulin; Syk, spleen tyrosine kinase.
Pathophysiology and targeted therapy for post-HSCT immune-mediated anemia. Post-HSCT anemia is multifactorial as a result of new auto- or alloantibodies in combination with T cell– and cytokine-mediated inflammatory processes. There are no formal guidelines in management. Several therapeutic approaches that target different pathophysiologic aspects of post-HSCT immune-mediated anemia are outlined. DLI, donor lymphocyte infusion; IVIG, IV immunoglobulin; Syk, spleen tyrosine kinase.
The complexity of post-HSCT immune-mediated anemia requires close collaboration between transfusion medicine, hematology, and transplantation services. Detailed knowledge about recipient/donor blood group phenotypes and the temporal association between transplantation and hemolysis can help identify the underlying mechanism of hemolysis and direct appropriate management. This review summarizes the literature on post-HSCT immune-mediated anemia to provide insight into its pathophysiology, clinical manifestations, and preventive and proposed therapeutic strategies.
ABO mismatch
ABO mismatch is divided into major, minor, and bidirectional categories (Table 1).
Major ABO mismatch
Major ABO mismatch is characterized by the presence of naturally existing host isohemagglutinins against the corresponding carbohydrate ABO antigens of the donor RBCs. The most common scenario is when a group O recipient receives a non-O graft; less commonly, a group A or B recipient may receive a B or A graft, respectively, or an AB graft. The naturally occurring isohemagglutinins in recipients can attack donor RBC ABO antigens early after stem cell infusion or can affect erythroid precursors later in the post-HSCT course.
ABO isohemagglutinins are immunoglobulin M (IgM) antibodies that can robustly activate the classical complement pathway.15 Through formation of the membrane attack complex, major ABO incompatibility may cause acute hemolytic transfusion reactions at the time of stem cell infusion, especially when the graft contains a significant volume of RBCs, such as in unmanipulated bone marrow (BM) grafts.16 Furthermore, recipient isohemagglutinins against donor-derived erythroid precursor cells can lead to reticulocytopenia with subsequent PRCA, which is seen in 8% to 26% of major ABO-mismatched HSCTs.17 PRCA usually occurs ∼30 to 90 days post-HSCT and is characterized by an absence of erythroid precursors in otherwise normal BM.18 Because a majority of recipients clear the donor-specific isohemagglutinins within 120 days, the development of PRCA has been attributed to residual recipient plasma cells that continue to produce isohemagglutinins (Figure 3).4 Patients with PRCA can be transfusion dependent for months, become iron overloaded, and develop alloimmunization to other blood group antigens.19 ABO antigens are also expressed on granulocytes and platelets; therefore, major ABO mismatch can also lead to prolonged neutropenia and thrombocytopenia, resulting in severe infections and catastrophic bleeding.20
Whether major ABO mismatch can affect HSCT outcomes, such as survival, relapse, and GVHD, is controversial (Table 2). A single-center study of 1502 patients undergoing HSCT with different graft sources, including peripheral blood (PB), BM or cord blood, showed that ABO mismatch was not associated with neutrophil or platelet engraftment delay, incidence of acute or chronic GVHD, OS, or NRM, regardless of graft source.21 A meta-analysis of 7 cohort studies showed that OS was not affected by ABO matching status.22 In contrast, in a Center for International Blood and Marrow Transplant Research study that included 5179 patients, major ABO mismatch was associated with decreased OS (hazard ratio [HR], 1.19; 95% confidence interval [CI], 1.19-1.31; P < .001) and increased NRM (HR, 1.23; 95% CI, 1.08-1.4; P = .002).23 In a European Blood and Marrow Transplant Acute Leukemia Working Group Registry study that included 837 patients who underwent haploidentical HSCT, major ABO mismatch was associated with an inferior day-100 engraftment rate, and in BM HSCT, it was associated with inferior OS.24
Selected studies evaluating ABO compatibility and transplantation outcomes (2000-2020)
| Reference | Year | ABO match, n (%)* | RIC, n (%) | Related donor, n (%) | BM graft, n (%)* | GVHD prophylaxis | Engraftment | GVHD rate | Relapse rate | NRM | OS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 16 | 2000 | 083 (53)65 (41)10 (6) | None | All related | —62 (74.7)46 (70.8)7 (70.0) | CNI + MTX | NS | No data | No data | No data | No data |
| 118 | 2001 | 2860 (41)1670 (24)1802 (26)587 (8) | No data | All matched unrelated | All BM | Various regimens | NS | NS | No data | No data | NS |
| 119 | 2005 | 2108 (68)451 (21)430 (14)114 (4) | 258 (12)46 (10)54 (13)17 (15) | All related | All BM | CNI + MTX | Delayed engraftment in major ABO incompatibility | NS | NS | NS | NS |
| 120 | 2007 | 121 (56)40 (19)40 (19)15 (7) | 0.5% of all patients | 86 (71)No data17 (43)No data | No data | CNI ± MTX; MTX alone | NS | NS | NS | NS | NS |
| 121 | 2008 | Major 205 (18.5)Minor 187 (17) | All patients | 932 (84) of all patients | 213 (19) of all patients | CNI only in 430 (39%) | No data | Higher in minor ABO incompatibility | No data | No data | Worse in minor ABO incompatibility |
| 122 | 2008 | 2820 (47)1834 (31)1202 (20)143 (2) | 348 (12)152 (8)136 (11)30 (21) | No data | No data | CNI + MTX | Delayed engraftment in major ABO incompatibility | Higher in major and minor ABO incompatibility | NS | Higher in major and minor ABO incompatibility | Worse in major and minor ABO incompatibility |
| 43 | 2009 | 58 (38)30 (19)44 (29)22 (14) | 17 (29)8 (27)15 (34)16 (73) | All matched unrelated | 27 (47)8 (27)22 (50)9 (41) | CNI + MTX/MMF; 10% with CNI only | No data | Higher acute GVHD rate in minor ABO incompatibility | NS | NS | NS |
| 23 (Stanford) | 2015 | 1053 (61)297 (17)309 (18)78 (4) | 526 (30) of all patients | 1303 (75) of all patients | 727 (42) of all patients | Various | No data | NS | NS | Higher in minor ABO incompatibility | Worse in minor ABO incompatibility, especially in BM grafts |
| CIBMTR(B-cell lymphoma) | 240 (59)73 (18)73 (18)22 (5) | 238 (55) of all patients | 330 (76) of all patients | None | Various | No data | NS | NS | Higher in minor ABO incompatibility | Worse in minor ABO incompatibility | |
| CIBMTR (AML or MDS) | 2608 (50)1084 (21)977 (19)311 (6) | 1448 (28) of all patients | 2079 (40) of all patients | 2333 (45) of all patients | Various | No data | NS | NS | Higher in major ABO incompatibility | Worse in major ABO incompatibility | |
| 42 | 2016 | 252 (49)105 (21)117 (23)38 (7) | 192 (76)78 (74)86 (74)30 (79) | 108 (43)25 (24)32 (27)7 (18) | None | No data | Delayed PLT engraftment in major ABO incompatibility | NS | NS | No data | NS |
| 123 (mismatched unrelated donor, AML) | 2017 | 349 (40)215 (25)241 (28)71 (8) | 193 (55)122 (57)154 (64)38 (54) | None | None | CNI-based in 87% of all patients | NS | Lower grade 2-4 acute GVHD rate in major ABO incompatibility | NS | NS | NS |
| 24 (haploidentical, AML) | 2017 | 522 (63)127 (15)150 (18)38 (5) | 215 (41)45 (35)68 (45)12 (32) | All haploidentical | 279 (53)76 (60)83 (55)19 (55) | No data | Delayed engraftment in major ABO incompatibility | Higher grade 2-4 acute GVHD in bidirectional | NS | NS | Worse in major ABO incompatibility + BM graft |
| 21 | 2017 | 704 (47)324 (22)372 (25)102 (7) | 301 (43)128 (40)151 (41)54 (53) | 372 (53)101 (31)88 (24)23 (23) | 296 (42)87 (27)73 (17)19 (19) | No data | Delayed neutrophil engraftment only in bidirectional with umbilical cord blood graft | NS | NS | NS | NS |
| 41 (severe aplastic anemia) | 2020 | 114 (57)47 (24)38 (19)0 | All patients | All haploidentical | Both BM and PB in all patients | CNI + MMF | NS | Grade 3-4 acute GVHD more common in minor ABO incompatibility | No data | No data | NS |
| 124 | 2020 | 590 (59.0)164 (16.4)191 (19.1)55 (5.5) | 110 (17)32 (20)35 (18)18 (33) | 531 (90)124 (76)145 (76)34 (62) | 189 (32)33 (20)41 (22)8 (15) | CNI + MTX | Neutrophil engraftment delayed in mismatched groups | NS | NS | NS | NS |
| Reference | Year | ABO match, n (%)* | RIC, n (%) | Related donor, n (%) | BM graft, n (%)* | GVHD prophylaxis | Engraftment | GVHD rate | Relapse rate | NRM | OS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 16 | 2000 | 083 (53)65 (41)10 (6) | None | All related | —62 (74.7)46 (70.8)7 (70.0) | CNI + MTX | NS | No data | No data | No data | No data |
| 118 | 2001 | 2860 (41)1670 (24)1802 (26)587 (8) | No data | All matched unrelated | All BM | Various regimens | NS | NS | No data | No data | NS |
| 119 | 2005 | 2108 (68)451 (21)430 (14)114 (4) | 258 (12)46 (10)54 (13)17 (15) | All related | All BM | CNI + MTX | Delayed engraftment in major ABO incompatibility | NS | NS | NS | NS |
| 120 | 2007 | 121 (56)40 (19)40 (19)15 (7) | 0.5% of all patients | 86 (71)No data17 (43)No data | No data | CNI ± MTX; MTX alone | NS | NS | NS | NS | NS |
| 121 | 2008 | Major 205 (18.5)Minor 187 (17) | All patients | 932 (84) of all patients | 213 (19) of all patients | CNI only in 430 (39%) | No data | Higher in minor ABO incompatibility | No data | No data | Worse in minor ABO incompatibility |
| 122 | 2008 | 2820 (47)1834 (31)1202 (20)143 (2) | 348 (12)152 (8)136 (11)30 (21) | No data | No data | CNI + MTX | Delayed engraftment in major ABO incompatibility | Higher in major and minor ABO incompatibility | NS | Higher in major and minor ABO incompatibility | Worse in major and minor ABO incompatibility |
| 43 | 2009 | 58 (38)30 (19)44 (29)22 (14) | 17 (29)8 (27)15 (34)16 (73) | All matched unrelated | 27 (47)8 (27)22 (50)9 (41) | CNI + MTX/MMF; 10% with CNI only | No data | Higher acute GVHD rate in minor ABO incompatibility | NS | NS | NS |
| 23 (Stanford) | 2015 | 1053 (61)297 (17)309 (18)78 (4) | 526 (30) of all patients | 1303 (75) of all patients | 727 (42) of all patients | Various | No data | NS | NS | Higher in minor ABO incompatibility | Worse in minor ABO incompatibility, especially in BM grafts |
| CIBMTR(B-cell lymphoma) | 240 (59)73 (18)73 (18)22 (5) | 238 (55) of all patients | 330 (76) of all patients | None | Various | No data | NS | NS | Higher in minor ABO incompatibility | Worse in minor ABO incompatibility | |
| CIBMTR (AML or MDS) | 2608 (50)1084 (21)977 (19)311 (6) | 1448 (28) of all patients | 2079 (40) of all patients | 2333 (45) of all patients | Various | No data | NS | NS | Higher in major ABO incompatibility | Worse in major ABO incompatibility | |
| 42 | 2016 | 252 (49)105 (21)117 (23)38 (7) | 192 (76)78 (74)86 (74)30 (79) | 108 (43)25 (24)32 (27)7 (18) | None | No data | Delayed PLT engraftment in major ABO incompatibility | NS | NS | No data | NS |
| 123 (mismatched unrelated donor, AML) | 2017 | 349 (40)215 (25)241 (28)71 (8) | 193 (55)122 (57)154 (64)38 (54) | None | None | CNI-based in 87% of all patients | NS | Lower grade 2-4 acute GVHD rate in major ABO incompatibility | NS | NS | NS |
| 24 (haploidentical, AML) | 2017 | 522 (63)127 (15)150 (18)38 (5) | 215 (41)45 (35)68 (45)12 (32) | All haploidentical | 279 (53)76 (60)83 (55)19 (55) | No data | Delayed engraftment in major ABO incompatibility | Higher grade 2-4 acute GVHD in bidirectional | NS | NS | Worse in major ABO incompatibility + BM graft |
| 21 | 2017 | 704 (47)324 (22)372 (25)102 (7) | 301 (43)128 (40)151 (41)54 (53) | 372 (53)101 (31)88 (24)23 (23) | 296 (42)87 (27)73 (17)19 (19) | No data | Delayed neutrophil engraftment only in bidirectional with umbilical cord blood graft | NS | NS | NS | NS |
| 41 (severe aplastic anemia) | 2020 | 114 (57)47 (24)38 (19)0 | All patients | All haploidentical | Both BM and PB in all patients | CNI + MMF | NS | Grade 3-4 acute GVHD more common in minor ABO incompatibility | No data | No data | NS |
| 124 | 2020 | 590 (59.0)164 (16.4)191 (19.1)55 (5.5) | 110 (17)32 (20)35 (18)18 (33) | 531 (90)124 (76)145 (76)34 (62) | 189 (32)33 (20)41 (22)8 (15) | CNI + MTX | Neutrophil engraftment delayed in mismatched groups | NS | NS | NS | NS |
AML, acute myeloid leukemia; CIBMTR, Center for International Blood and Marrow Transplant Research; CNI, calcineurin inhibitor; MDS, myelodysplastic syndrome; MMF, mycophenolate mofetil; MTX, methotrexate; NRM, nonrelapse mortality; NS, not significant; OS, overall survival; PLT, platelets; RIC, reduced-intensity conditioning.
Data given in the following order: matched, major, minor, bidirectional.
Patients with ABO mismatch may have lower rates of complications if they receive myeloablative (MA) conditioning.25 One study showed that fludarabine/cyclophosphamide conditioning, compared with a total-body irradiation–based MA regimen, was associated with delayed full donor erythroid chimerism by a median of 74 days and increased risk of PRCA. The clearance of antidonor isohemagglutinins was faster in the latter.25 Other studies have also shown impaired HSCT outcomes in ABO-mismatched HSCTs involving RIC.26,27 ABO mismatch was associated with a 1.7-fold increased risk of extensive chronic GVHD in a cohort of 594 patients who underwent alemtuzumab-based RIC HSCT.27
Preventive strategies for immunohematologic complications.
Although ABO matching is not required for donor selection per current guidelines, it is worth taking into consideration when multiple suitable donors are available.28 Besides using MA regimens when feasible, strategies to prevent complications in major ABO mismatch include graft RBC reduction and removal of host isohemagglutinins by therapeutic plasma exchange before HSCT.29-31 In recipients with antidonor isohemagglutinin titers ≥1:32, RBC depletion should be performed to ensure infused RBCs <20 mL. When titers are ≤1:16, RBC depletion is generally not required.3
Because ABO antigens can be secreted into the plasma, pretransplantation immunoabsorption (ie, infusion of donor-type fresh frozen plasma [FFP] positive in A or B antigens to neutralize respective isohemagglutinins) is another approach to prevent complications. In a single-center study in Singapore, 79 of 99 major or bidirectional ABO-mismatched recipients were treated with donor FFP from day −5 to −2 before transplantation, including 70 patients with isohemagglutinin titers >1:32. Those with a titer level ≤1:32 at the time of stem cell infusion did not develop acute hemolysis. Three patients developed PRCA irrespective of pre- or post-FFP isohemagglutinin levels.32 In another single-center study from Australia, 75 of 110 major or bidirectional ABO-mismatched recipients had isohemagglutinin titers >1:32 and received FFP with or without plasma exchange before HSCT. The incidence of PRCA was 5%. The risk of PRCA or delayed RBC engraftment was significantly higher in patients with pretransplantation isohemagglutinin titers ≥1:128.17
Donor-type RBC transfusion is another immunoabsorption method.33,34 In a single-center study in thalassemia, 20 of 55 major or bidirectional ABO-mismatched recipients with isohemagglutinin titers ≥1:32 received small incremental doses of uncrossmatched donor RBCs during conditioning; 12 had mild hemolysis, and none had severe hemolysis or anaphylaxis. In patients who had no hemolytic reaction at the last transfusion or who had titers <1:32, BM grafts were infused without RBC depletion. No hemolytic reactions were observed post-HSCT, and all patients experienced engraftment without delay.33 Despite these successes, immunoabsorption requires high expertise in transfusion medicine and close patient monitoring, and it remains to be validated in larger studies.
Minor ABO mismatch
Minor ABO mismatch is characterized by the interaction of donor-derived isohemagglutinins with corresponding recipient RBC antigens. These isohemagglutinins may be infused along with the stem cell products, causing acute hemolysis of recipient RBCs. Alternatively, passenger B-lymphocytes from the donor graft may proliferate and secrete isohemagglutinins and other RBC antibodies, causing clinically significantly delayed hemolysis called PLS (Figure 4).35 PLS typically occurs 1 to 2 weeks post-HSCT, more frequently after PB than BM HSCT, because the former contains a higher concentration of lymphocytes.35,36 RIC is associated with a higher risk of PLS because recipient antigens may be left intact and subsequently stimulate donor B cells.37,38 A cyclosporine-only GVHD prophylactic regimen has been associated with higher risks of immunohematologic complications because cyclosporine boosts B-cell antibody production.2,39
The impact of minor ABO mismatch on HSCT outcomes is also controversial (Table 1). Some studies suggest that minor ABO mismatch is a risk factor for acute GVHD, decreased OS, and increased NRM.23,40-42 One study found it was associated with a threefold increased risk of grade 2 to 4 acute GVHD and fourfold increased risk of grade 3 to 4 acute GVHD.43 In a 1737-patient cohort at Stanford University between 1986 and 2011, minor ABO mismatch was an independent risk factor for OS, with an HR of 1.56 (95% CI, 1.05-2.05; P = .001).23 In a Center for International Blood and Marrow Transplant Research analysis of patients with lymphoma receiving PB grafts, minor ABO mismatch was associated with reduced OS and higher NRM (HR, 1.55 and 1.72; P = .021 and .03, respectively); however, these associations were not observed in patients with acute myeloid leukemia or myelodysplastic syndrome.23
Preventive strategies for immunohematologic complications.
In minor ABO-mismatched HSCT, immediate hemolysis can be prevented by graft plasma volume reduction; this does not prevent PLS because it does not affect the B-cell component in the graft.44 There is a scarcity of literature on how to prevent PLS. Patients need close monitoring for hemolysis, especially at ∼1 to 2 weeks post-HSCT when PLS may occur. A direct antiglobulin test may assist in early detection of subclinical hemolysis. It is often checked once or twice weekly according to institutional protocols. Maintaining higher hemoglobin levels than the normal transfusion threshold is preferred, especially during the high-risk period, to avoid nadir in case severe or life-threatening PLS occurs. Collaborative efforts between transfusion and transplantation services to ensure the awareness of minor ABO mismatch and transfusion-compatible RBC components peritransplantation is critical.
Bidirectional ABO mismatch
Bidirectional mismatch occurs when the donor/recipient pair has both a major and minor ABO mismatch, such as type A donor/type B recipient or vice versa. The recipient is at risk of complications associated with both directions of incompatibility (Table 1), and preventive methods should address both accordingly.
Non-ABO blood group incompatibility
Unlike ABO isohemagglutinins, which exist in the absence of previous exposure, alloimmunization to protein antigens occurs only after exposure. The incidence of alloimmunization after transfusion is 2% to 6% and most commonly involves the Rhesus (Rh), Kell (K), and Kidd (Jk) blood groups.45-47 RhD antigen is the most potent non-ABO antigen, followed by K antigen. Patients with a history of alloimmunization are at higher risk of developing additional alloantibodies.48 Although rare, non-ABO blood group alloantibodies may result in poor HSCT outcomes.49,50 Complement cascade is implicated in immune-mediated anemia in some of the non-ABO–mismatched HSCT. For example, the IgG or IgM antibodies against Jk antigens are able to fix complements and induce intra- or extravascular hemolysis.51 Mismatch of the Jk antigen system has been reported to cause severe hemolytic anemia or PLS.8,52
RBC alloimmunization resulting from non-ABO mismatch could originate from the donor, recipient, or both, depending on the temporal relationship between antibody development and chimerism status. Donor-derived alloantibodies include the preexisting antibodies in the graft and new antibodies produced by passenger or engrafted lymphocytes. Recipient-derived alloantibodies include preexisting antibodies and antibodies produced by residual lymphocytes and plasma cells.
Post-HSCT, a majority of the de novo non-ABO alloantibodies develop within the first month.8 PLS can also occur in non-ABO blood group–mismatched HSCT, usually involving RhD, E, s, Jkb, or Jka.52-54 Table 3 outlines previously described clinically significant non-ABO blood group systems associated with alloimmunization after HSCT.
Non-ABO blood group systems with potential to cause clinically significant alloimmunization after HCT
Patients receiving chronic transfusion therapy before HSCT are generally at higher risk of developing auto- and alloimmunization. Patients with sickle cell disease have high rates of RBC alloimmunization (23.8% to 45.7%) despite leukoreduction and prophylactic antigen matching.55,56 Frequent transfusions, lack of phenotypically matched products, inadequate alloantibody testing, and presence of antigen variations, especially in donors of African descent, increase the risk of alloimmunization.57 In 1 cohort, RBC antibodies were detected in 9 (15%) of 61 patients post-HSCT, including 3 patients with preformed and 6 with new antibodies, and 4 patients developed reticulocytopenia or hemolysis.58 In a cohort of patients undergoing matched-sibling HSCT without MA conditioning, 31% (11 of 31) had a history of RBC alloantibodies, which was correlated with decreased donor T-cell chimerism at 1 year compared with patients without alloimmunization (median, 24% vs 55%; P = .035).59 As the number of HSCTs in sickle cell disease is increasing, it remains a challenge to prevent and treat RBC alloimmunization peri-HSCT.
Thalassemia is another hemoglobinopathy that requires chronic transfusion therapy. Alloantibodies occur in 5% to 30% of patients with thalassemia and are more common after splenectomy, long duration of treatment, and frequent transfusions.60-63 The dominant antibodies are against the Rh and K groups, each comprising 20% to 30% of the antibodies.63 When prophylactically transfusing Rh (D, C, and E) antigen– and K antigen–matched RBC units, alloantibodies were still detected in 32.5% of patients, with an alloimmunization rate of 0.26 antibodies per 100 units, and 72.5% of antibodies were directed against Rh.62 There are limited data on how non-ABO alloantibodies affect HSCT outcomes in patients with thalassemia.
Chronic granulomatous disease can coexist with the McLeod phenotype (ie, reduced expression of K blood group antigens and absence of XK protein on RBCs). This is due to the genetic proximity between the cytochrome B β subunit gene and the XK gene. Hönig et al64 reported a case of successful HSCT in a patient with chronic granulomatous disease/McLeod phenotype with anti-K and anti-Kx alloantibodies. The patient received rituximab, antithymoglobin, MA conditioning, and a K− HLA-matched unrelated graft. The patient had an uneventful HSCT course with prompt engraftment. The anti-K alloantibodies remained detectable until 20 months post-HCT.64 Another patient received MA conditioning with busulfan, cyclophosphamide, and antithymoglobin, followed by a K− BM graft. He developed refractory severe hemolytic anemia after day +100 as a result of anti-Kx and anti-K and was found to have low donor T-cell chimerism, despite B-cell and erythroid conversion. His hemolytic anemia resolved after 3 DLI.65
Outside of the chronically transfused population, the rate of non-ABO alloimmunization is reportedly low.7,8,10 De la Rubia et al10 reported a rate of 3.7% (8 of 217) of patients developing new non-ABO alloantibodies post-HCT, 2 of whom developed severe immune hemolytic anemia early after HCT; recipient age and ABO incompatibility were associated with the development of non-ABO alloantibodies. Ting et al7 reported a rate of 8.7% (13 of 150) of patients experiencing new RBC alloantibody production from 12 days to 11 months post-HCT. In another retrospective study, the incidence of alloimmune hemolysis post-HCT involving non-ABO antigens was 1% (5 of 427 patients).8 When alloantibodies are detected promptly, these complications may be prevented.8,66 In 1 case, a recipient with anti-Jka antibodies received fludarabine/melphalan conditioning with rituximab, followed by Jka+ donor PB stem cells without adverse events.66 Complications related to non-ABO antigen incompatibility are expected to increase given the progressive success in HSCT and its wide use in malignant and nonmalignant diseases.67
Laboratory investigations in non-ABO blood group–mismatched HSCT
Immunohematologic complications caused by non-ABO blood group mismatch can be underdiagnosed because of inadequate phenotyping of the donor-recipient RBCs. In the absence of known preexisting alloantibodies in the recipient, testing beyond the ABO and Rh blood groups is generally not considered before transplantation. Some alloantibodies may be not detected, such as the Jka/Jkb antibodies, because of fluctuation of the titers.8 Jk antibodies are known to have an amnestic response such that a patient may test negative initially but have a subsequent reemergence of antibodies later in the post-HSCT course.
Extended serologic RBC phenotype matching is being performed for patients receiving frequent transfusions in order to provide appropriate antigen-negative blood products. Despite this, alloantibodies can still form and lead to additional challenges in transfusion.68 Extended genotype RBC matching can detect and predict minor variations in RBC antigens that are hard to identify serologically; it is increasingly used in clinical practice.69,70 Real-time polymerase chain reaction is another method for quick RBC phenotyping in HSCT, especially when donor-recipient myeloid chimerism status is dynamically changing.71
Anemia resulting from autoimmunity
AIHA occurs in 4% to 6% of those undergoing HSCT.72,73 In 1 study of T cell–depleted haploidentical HSCT in patients with severe combined immunodeficiency, the incidence of AIHA was 19.5%.74 Patients post–second HSCT have higher incidence and earlier presentation of AIHA than those post–first HSCT.75 AIHA may occur alone or in conjunction with other immune-mediated cytopenias.73 RBC phenotyping of the donor and recipient, determined before HSCT, can help differentiate autoimmune-mediated anemia from alloimmunity.76 In different reports, median time of onset ranges from 4 to 10 months post-HSCT.75 Transplantation for nonmalignant disease is the most consistent risk factor for post-HSCT AIHA in the literature; other factors, such as unrelated donor, acute or chronic GVHD, cytomegalovirus activation, and alemtuzumab-containing conditioning regimen, have been reported, but reports are inconsistent.72,76-78
The pathophysiology of AIHA post-HSCT has not been fully elucidated, but it may be related to preformed autoantibodies and dysregulated immune tolerance. In the chronically transfused population, autoantibodies against RBCs have been reported in 7% to 25% of patients; a majority of the autoantibodies are transient, but some may lead to clinically significant hemolysis.79 Patients who develop AIHA are severely lymphopenic, with low numbers of regulatory T cells; delayed T-cell immune reconstitution, resulting from either in vivo or ex vivo T-cell depletion, may allow autoreactive B cells to activate and expand without regulation.74,80 In contrast, flowcytometry and cytokine analysis of patients with AIHA post-HSCT identified a decreased CD3+CD8+ T-cell ratio and a T-helper cell 2–related cytokine profile compared with control patients.76 Patients who develop AIHA are at a higher risk for developing alloimmunity.
Treatment
Mild to moderate cases of hemolytic anemia, including PLS, are self-limited and are usually treated with simple transfusions of antigen-negative RBCs; however, finding the appropriate RBC unit can be challenging when multiple alloantibodies or antibodies to high-frequency antigens are present.81 Approximately 50% of PRCA cases resolve spontaneously within 100 to 200 days post-HSCT, but in cases of prolonged anemia dependent on chronic frequent transfusions, treatment is necessary to prevent complications such as iron overload.18,82-84
Most of the therapeutic options for post-HSCT immune-mediated anemia are derived from AIHA, which does not have a licensed treatment itself (Figure 2).80 Based on mechanism of action, the options can be categorized into immunosuppression, which includes the use of systemic steroids, IV immunoglobulin, rituximab, cyclophosphamide, azathioprine, and splenectomy in the severe cases; immunoabsorption; RBC or plasma exchange; and stimulation of erythropoiesis using erythropoietin or thrombopoietin mimetics (Figure 1).85-89 For PRCA, the most commonly used therapies are rituximab, erythropoietin, and DLI (Figure 3).90
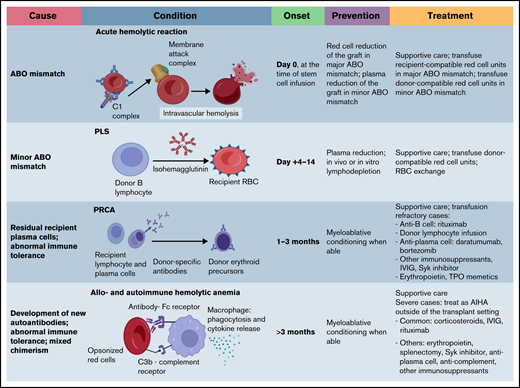
General approach for posttransplantation immune-mediated hemolysis. The graph outlines underlying mechanisms of posttransplantation hemolysis, timing posttransplantation, preventive methods, and treatment strategies. Note there is no consensus or guideline on how to manage posttransplantation immune-mediated anemia. The management approaches listed are based on expert opinions and available literature. AIHA, autoimmune hemolytic anemia; IVIG, IV immunoglobulin; TPO, thrombopoietin.
General approach for posttransplantation immune-mediated hemolysis. The graph outlines underlying mechanisms of posttransplantation hemolysis, timing posttransplantation, preventive methods, and treatment strategies. Note there is no consensus or guideline on how to manage posttransplantation immune-mediated anemia. The management approaches listed are based on expert opinions and available literature. AIHA, autoimmune hemolytic anemia; IVIG, IV immunoglobulin; TPO, thrombopoietin.
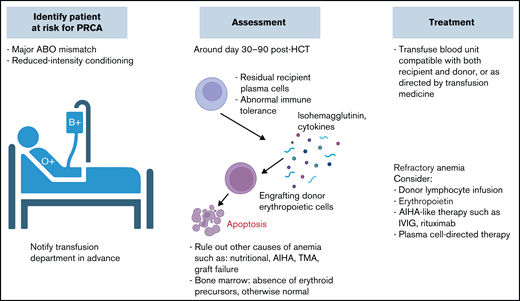
PRCA. PRCA is more frequently encountered in major ABO mismatch during the first 1 to 3 months posttransplantation. Management approach is similar to PLS, with frequent monitoring and RBC transfusion support. Other pharmacologic interventions can be considered in refractory cases. CBC, complete blood count; Hb, hemoglobin; IVIG, IV immunoglobulin; TMA, thrombotic microangiopathy.
PRCA. PRCA is more frequently encountered in major ABO mismatch during the first 1 to 3 months posttransplantation. Management approach is similar to PLS, with frequent monitoring and RBC transfusion support. Other pharmacologic interventions can be considered in refractory cases. CBC, complete blood count; Hb, hemoglobin; IVIG, IV immunoglobulin; TMA, thrombotic microangiopathy.
There are reports of efficacy using anti-CD20 monoclonal antibodies such as rituximab to treat PRCA.91 Because B cells are responsible for isohemagglutinins or alloantibody production in PLS, rituximab can also be used in severe cases of PLS (Figure 4).92 Other immunosuppressive treatments reported to work in refractory cases include abatacept, a fusion protein that blocks interaction between T cells and antigen-presenting cells.93
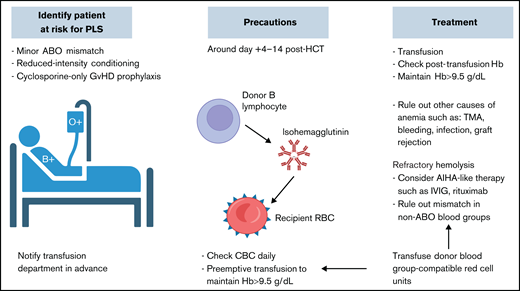
PLS. Proactive approach with identifying patients at risk before transplantation, alerting the blood bank service, conducting frequent count monitoring, and providing transfusion support in the first 2 weeks posttransplantation is key for better clinical outcomes. Hb, hemoglobin; IVIG, IV immunoglobulin; TMA, thrombotic microangiopathy.
PLS. Proactive approach with identifying patients at risk before transplantation, alerting the blood bank service, conducting frequent count monitoring, and providing transfusion support in the first 2 weeks posttransplantation is key for better clinical outcomes. Hb, hemoglobin; IVIG, IV immunoglobulin; TMA, thrombotic microangiopathy.
Erythropoietin-stimulating agents can increase RBC production and can be used as a therapeutic option in severe hemolytic anemia or PRCA post-HSCT.88,94 Eltrombopag is a thrombopoietin receptor agonist approved for the treatment of aplastic anemia. It has shown efficacy in refractory acquired PRCA outside the HSCT setting.95 Busca et al89 reported 2 cases of PRCA that were refractory to erythropoietin, plasma exchange, rituximab, and bortezomib; these patients achieved sustained PRCA remission with eltrombopag.
DLI and tapering of immunosuppressants represent unique therapeutic options in the HSCT setting. These methods may improve donor chimerism, eliminate recipient plasma cells, and reduce alloreactivity from either the donor or recipient direction. The efficacy of both methods in PRCA is ∼50% in case reports.83 In 1 case report, a patient with PRCA received DLI with a CD34+ stem cell boost and achieved normal blood counts after 2 months.96 However, in a recent multicenter retrospective study, rituximab, DLI, and erythropoietin had no impact on the resolution of PRCA.90 Mesenchymal stem cell infusion has also been used in refractory PRCA cases, with promising results.97,98
Plasma cells are a viable target for therapy because they may contribute to antibody production, especially in refractory hemolytic anemia and PRCA. Bortezomib was reported to be effective in 40% of cases in the literature.83 Compared with other plasma cell–targeted therapy, daratumumab has a more appealing safety profile and mechanism of action.99,100 A few case reports have noted its efficacy in refractory post-HSCT AIHA and PRCA.101-103 Daratumumab may have efficacy in other forms of post-HCT immune-mediated cytopenia, such as thrombocytopenia or Evan’s syndrome.104 We previously reported durable response after daratumumab in a patient with posttransplantation thrombocytopenia.105 Daratumumab impairs RBC crossmatch testing, which necessitates transfusing K− units posttreatment, unless the RBC phenotype is identified pretreatment.106 However, daratumumab is not always effective in post-HSCT immune-mediated cytopenia, likely because of the suppressive effect of daratumumab on CD38+ regulatory T cells.107,108
A novel modulatory therapy that is worth testing is the Syk inhibitor fostamatinib. Its active metabolite, R406, was shown to reduce antibody-mediated platelet destruction. It was US Food and Drug Administration approved in April 2018 for patients with chronic immune thrombocytopenia for whom 1 line of therapy failed. Additionally, it is currently being studied in a phase 3 clinical trial for patients with warm AIHA (registered at www.clinicaltrials.gov as #NCT03764618) and in an early-phase chronic GVHD trial (#NCT02611063). The Syk pathway is critical for B-cell activation and proliferation; activated B cells may trigger T-cell activation and cytokine production.109,110 Preclinical reports in xenograft models using PB mononuclear cells from patients with active chronic GVHD showed that the Syk inhibitor augments B-cell apoptosis while not affecting normal T-cell function.111
Therapies targeting the complement pathway are being more frequently used in immune-mediated cytopenia. Vo et al112 used eculizumab, a complement C5 inhibitor, to treat heavily alloimmunized patients with platelet transfusion refractoriness. Four of the 10 patients enrolled overcame platelet transfusion refractoriness with 1 dose of eculizumab. Eculizumab has shown efficacy in warm antibody AIHA and cold agglutinin disease.113,114 In post-HSCT AIHA, eculizumab was effective in 1 of the 3 cases reported in the literature.72 Several other complement inhibitors, such as pegcetacoplan and sutimlimab, are actively being investigated in complement-mediated hemolytic anemia.115,116 These agents may be viable options for refractory hemolytic anemia post-HSCT.
Therapies carry their own risks. Infection is the most concerning complication in patients receiving immunosuppression. In 1 study analyzing the outcome of 46 patients with PRCA, 22 received treatment other than supportive care, and 7 died as a result of infection.18 IV immunoglobulin, rituximab, and plasma exchange may cause anaphylactic reactions. DLI may lead to higher incidence of GVHD.117 Therefore, careful evaluation of the risks and benefits before initiation of therapy is crucial, requiring multidisciplinary collaboration between transfusion medicine, hematology, and HSCT providers.
Conclusions
Donor-recipient RBC antigen mismatch is commonly encountered and results in clinically significant immunohematologic events, complicating the posttransplantation course. ABO phenotype is part of the donor-recipient pretransplantation workup to plan transfusion needs, clinical laboratory monitoring, and possible graft manipulations. Donor-recipient extended phenotyping for non-ABO antigens, such as Rh, K, and Jk, should be considered in high-risk groups, especially those receiving chronic transfusion therapy. HSCT across the barrier of RBC antigen mismatch may lead to acute hemolysis, PLS, and PRCA. Early detection of hemolysis, supportive care with antigen-negative or matched RBC transfusion, and appropriate interventions such as immunosuppression are necessary to improve clinical outcomes. Active collaboration between transfusion and transplantation services might help with early identification of clinically significant antibodies and prompt necessary treatment interventions.
Acknowledgments
The authors extend their gratitude to the patients reported in this review and thank Charles Bolan for his contribution and review of this manuscript.
Authorship
Contribution: Y.M. and Y.P. provided conception and design and wrote, reviewed, and/or revised the manuscript; S.S.K. reviewed and revised the manuscript; and S.A. and R.C. wrote and revised the manuscript and provided expert guidance.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yazan Migdady, 3181 SW Sam Jackson Park Rd, Portland, OR 97239; e-mail: migdady@ohsu.edu.

