

Key Points
A missense mutation in LCP1, an actin-bundling protein, results in severe neutropenia, migration defects, and susceptibility to infection.
LCP1 mutation causes actin dysregulation with increased F-actin, G2/M cell cycle arrest, dysplastic cells, and nuclear mislocalization.

Abstract
Neutrophils migrate into inflamed tissue, engage in phagocytosis, and clear pathogens or apoptotic cells. These processes require well-coordinated events involving the actin cytoskeleton. We describe a child with severe neutropenia and episodes of soft tissue infections and pneumonia. Bone marrow examination showed granulocytic hypoplasia with dysplasia. Whole-exome sequencing revealed a de novo heterozygous missense mutation in LCP1, which encodes the F-actin–binding protein Lymphocyte Cytosolic Protein 1. To determine its pathophysiological significance, we stably transduced cells with doxycycline-inducible wild-type LCP1 and LCP1 I232F lentiviral constructs. We observed dysplastic granulocytic 32D cells expressing LCP1 I232F cells. These cells showed decreased proliferation without a block in differentiation. In addition, expression of LCP1 I232F resulted in a cell cycle arrest at the G2/M phase, but it did not lead to increased levels of genes involved in apoptosis or the unfolded protein response. Both 32D and HeLa cells expressing mutant LCP1 displayed impaired cell motility and invasiveness. Flow cytometry showed increased F-actin. However, mutant LCP1-expressing 32D cells exhibited normal oxidative burst upon stimulation. Confocal imaging and subcellular fractionation revealed diffuse intracellular localization of LCP1, but only the mutant form was found in the nucleus. We conclude that LCP1 is a new gene involved in granulopoiesis, and the missense variant LCP1 I232F leads to neutropenia and granulocytic dysplasia with aberrant actin dynamics. Our work supports a model of neutropenia due to aberrant actin regulation.
Introduction
The inherited neutropenia syndromes are rare disorders that are phenotypically and genetically heterogeneous. Although >20 years have elapsed since the discovery of ELANE in cyclic neutropenia or severe congenital neutropenia (SCN),1,2 our understanding of the mechanisms leading to neutropenia remains limited. More than 20 different genes have been implicated in the pathogenesis of SCN, although 30% of patients have no identified genetic defect. Cellular stress has been implicated in the pathogenesis of inherited neutropenia, such as with endoplasmic reticulum stress,3-6 impaired endosomal trafficking,7,8 defective ribosomal biogenesis,9-11 and defects in bioenergetics.5 The role of cytoskeletal stress in neutropenia remains underinvestigated. X-linked SCN occurs secondarily to gain-of-function mutations in Wiskott-Aldrich syndrome protein (WASp). These mutations cause activation of Arp2/3 and actin polymerization, leading to failed cytokinesis, decreased proliferation, and increased apoptosis.12 However, the precise mechanism of how altered F-actin dynamics leads to neutropenia remains unclear.
With an integral role in cellular functions, actin and actin-interacting proteins are implicated in various immune and hematologic disorders.13-15 We present a pediatric patient with severe neutropenia and infections, who harbored a heterozygous missense mutation in LCP1, which encodes Lymphocyte Cytosolic Protein 1, an F-actin–binding protein. To elucidate how LCP1 I232F mutation leads to neutropenia, we stably transduced 32D and HeLa cells with complementary DNAs (cDNAs) for either wild-type (wt) LCP1 or LCP1 I232F under an inducible promoter. We discovered that the expression of LCP1 I232F resulted in increased F-actin polymerization and cell cycle arrest at the G2/M phase, leading to impaired cellular proliferation and dysplasia of myeloid cells.
Methods
Cell lines and culture conditions
Interleukin-3 (IL-3)–dependent murine myeloblast 32D cells (ATCC) were grown as described elsewhere.16 The LCP1 human cDNA was obtained from GeneCopoeia. The missense mutation c.694 A>T (p.I232F) was created by using the QuikChange Site-Directed Mutagenesis Kit (Thermo Fisher Scientific). The wtLCP1 and LCP1 I232F constructs were cloned into the pInducer20 vector system by using Gateway cloning (Thermo Fisher Scientific). To induce expression, media were supplemented with 1 µg/mL of doxycycline (MilliporeSigma). Doxycycline dose–response was performed to determine induction (supplemental Figure 1).
Cell count and morphology
Cell count and viability were performed by using trypan blue (Thermo Fisher Scientific) exclusion. To characterize cell morphology, cells were cytospun at a speed of 500 rpm for 5 minutes, fixed and then stained with Wright-Giemsa staining.
Immunoblotting
Cells were lysed either with radioimmunoprecipitation assay or NP40 cell lysis buffer (Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (MilliporeSigma) and sodium orthovanadate (MilliporeSigma). Lysates underwent electrophoresis and were transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% nonfat dry milk (Bio-Rad). Primary antibodies (supplemental Table 1) were applied in Tris-buffered saline Tween 20 with 5% nonfat dry milk overnight at 4°C, followed by incubation with a horseradish peroxidase–conjugated secondary antibody (Rockland Immunochemicals). For signal detection, the membranes were incubated with ECL Start Western Blot Detection Reagent (MilliporeSigma) followed by imaging on a ChemiDoc Imaging System (Bio-Rad).
Structural modeling
Homology modeling of LCP1–actin-binding domain 1 (ABD1) (sequence: 120-380) was performed by using the I-TASSER program.17,18 I-TASSER generated 5 model structures of LCP1-ABD1. All 5 models showed that the calponin homology domains 1 and 2 (CH1 and CH2, respectively) adopted the same 4-helix core architecture, and they were connected by an interdomain helix. To represent the LCP1-ABD1 model, we selected model 1, which had the highest score. Using the LCP1-ABD1 model, Ile232 was then mutated to Phe to obtain the LCP1-ABD1I232F model. To construct the complex model of LCP1-ABD1 and F-actin, we used the cryo–electron microscopy (cryo-EM) structure of LCP1-ABD2 and F-actin as the template and replaced one LCP1-ABD2 by either LCP1-ABD1 or LCP1-ABD1I232F after alignment of LCP1-ABD2-CH3 with LCP1-ABD1-CH1. To study the dynamic motion of LCP1-ABD1 and LCP1-ABD2, the homology models of LCP1-ABD1, LCP1-ABD1I232F, and LCP1-ABD2 from the cryo-EM structure were used to perform molecular dynamics simulations. The Amber 14SB force field parameters19 were used for the amino acids and the TIP3P20 model for water molecules. The GPU modification to PMEMD21 from Amber (version 18)22 was used in all molecular dynamics simulations. The system minimization and equilibration procedures followed our previous study.23 Figures were prepared by using the PyMOL program (www.pymol.org), VMD,24 and UCSF Chimera.25
Flow cytometry studies
All sample acquisition was performed by using a BD FACSVerse cytometer (BD Biosciences), and the data were analyzed by using FlowJo version 10.5.3 (FlowJo LLC). To study granulocytic differentiation, 32D cells were treated with 100 ng/mL of granulocyte colony-stimulating factor (G-CSF) for 8 days and labeled with fluorescein isothiocyanate–conjugated anti-mouse CD11b and phycoerythrin-conjugated anti-Ly6G antibodies (BD Biosciences). Apoptosis was analyzed by staining with Alexa Fluor 488–conjugated Annexin V and propidium iodide according to the manufacturer’s protocol (Dead Cell Apoptosis Kit, Thermo Fisher Scientific). For cell cycle analysis, 32D cells were subjected to overnight serum and cytokine starvation to induce G1 arrest. Fresh IL-3 and fetal bovine serum–containing medium was added, and the cells were harvested over a time course of 0 to 48 hours. The cells were washed in 1X phosphate-buffered saline (Thermo Fisher Scientific) and fixed in ice-cold 70% ethanol (Thermo Fisher Scientific). The fixed cells were washed with 0.1% Tween 20 (Thermo Fisher Scientific) in phosphate-buffered saline and then resuspended in DNA staining buffer (10 μg/mL propidium iodide and 250 μg/mL RNase A) and incubated for 15 minutes at 37°C. For quantification of intracellular F-actin, cells were permeabilized by using 0.5% Triton-X and probed by using fluorescein isothiocyanate–phalloidin (Santa Cruz Biotechnology). To measure oxidative burst, differentiated 32D cells (100 ng/μL G-CSF for 8 days) were treated with either dimethyl sulfoxide, N-formyl-Met-Leu-Phe (1 μM), or phorbol myristate acetate (100 nM) for 30 minutes. Levels of reactive oxygen species were measured by using dihydrorhodamine-123.26
Quantitative polymerase chain reaction–based gene expression analysis
Total RNA was isolated from cells by using TRIzol reagent (Thermo Fisher Scientific). Reverse transcription was performed by using the iScript II cDNA synthesis kit (Bio-Rad). The cDNA was used for real-time quantitative polymerase chain reaction to determine gene expression by using custom designed primers (supplemental Table 2). The quantitative polymerase chain reaction was performed by using Power Up SYBR Green Master Mix (Thermo Fisher Scientific) on a QuantStudio 3 system (Thermo Fisher Scientific). The gene expression analysis was performed by using the ΔΔ-CT method, with Actin as the reference gene and time 0 as the reference sample.
Migration assays
After HeLa cells grew to confluence in 6-well culture plates, uniform wound scratch lines were made within the marked location by using a pipette tip. The cells were allowed to migrate and close the wound in culture media with and without 1 μg/mL of doxycycline. The pictures of the scratch were captured at 0, 6, 24, and 48 hours by using a Zeiss SteREO Discovery V8 microscope (Zeiss). The wound gaps were measured by using ImageJ software (National Institutes of Health). Boyden chamber cell invasion was performed by using CytoSelect Colorimetric Cell Invasion Assay (Cell Biolabs). HeLa and 32D cells were starved in serum-free medium for 14 hours. Cells were plated in the upper chamber and allowed to migrate through a membrane filter for 48 hours with serum added to the lower chamber as chemoattractant. For HeLa cells, the migrated cells were stained by using crystal violet and counted under light microscope. The migrated cells were then harvested and lysed by using the extraction reagent, and the optical densities were measured at 560 nm by using the Synergy H1 Hybrid Multi-mode plate reader (BioTek). For 32D cells, the migrated cells were stained with 0.4% trypan blue (Thermo Fisher Scientific) and counted manually. Pictures were taken by using the Nuance Multispectral Imaging System.
Immunofluorescence and confocal microscopy
32D cells were cytospun onto the coverslips, whereas HeLa cells were grown on coverslips. Cells were fixed by using 4% formaldehyde for 15 minutes. The cells were permeabilized with 0.5% Triton-X for 15 minutes at room temperature and stained by using CruzFluor 488 phalloidin conjugate (Santa Cruz Biotechnology), LCP1 Alexa Fluor 594 (sc-133218), and 4′, 6-diamidino-2-phenylindole (5 μg/mL, Thermo Fisher Scientific). The coverslips were mounted on glass slides using Fluoroshield (MilliporeSigma) and imaged with a Leica SP8 Inverted Confocal Microscope.
Subcellular fractionation
A subcellular protein fractionation kit (Thermo Fisher Scientific) was used on 32D cells expressing wtLCP1 and LCP1 I232F into cytoplasmic, membrane, and nuclear fractions following the manufacturer’s protocol (Subcellular Protein Fractionation Kit for Cultured Cells, Thermo Fisher Scientific). Immunoblotting for subcellular marker proteins (supplemental Table 2) was used to determine fraction purity.
Statistical analysis
Prism 8.0 software (GraphPad Software) was used to perform descriptive and comparative analyses. The standard error of the mean was used. An unpaired two-way Student t test was used to detect significance between the means of 2 groups, whereas one- or two-way analysis of variance using Tukey’s multiple comparison test was used to compare multiple groups simultaneously. Statistical significance was defined as P value <.05.
Study approval
The study was approved by the Cleveland Clinic institutional review board.
Results
Identification of LCP1 mutation as a novel autosomal dominant cause of neutropenia
Our patient is a currently aged 7-year-old girl who presented at the age of 2 years with recurrent cellulitis of the leg and abdominal wall. She is non-dysmorphic and had normal neurodevelopmental milestones with no physical stigmata of inherited bone marrow failure syndromes. She was diagnosed with severe neutropenia with an absolute neutrophil count of 0.08 × 103/µL and monocytopenia with an absolute monocyte count of 0.08 × 103/µL. Her white blood count was 3.78 × 103/µL, hemoglobin 10.4 g/dL, and platelets 448 × 103/µL. The differential showed an absolute lymphocyte count of 3.33 × 103/µL and an eosinophil count of 0.3 × 103/µL. Initial evaluation of neutropenia included weekly complete blood cell counts to evaluate for periodicity, which were negative. Due to low CD3+CD8+ T-cell numbers in T-cell subset analysis, mitogen stimulation testing was performed, which revealed a normal lymphocyte proliferation response to phytohemagglutinin, concanavalin A, and pokeweed mitogen. Neutrophil antibody panel (Blood Center of Wisconsin) was negative for antineutrophil antibodies. Our patient remained severely neutropenic (Figure 1A). Results of the hematologic and immunologic workup are summarized in supplemental Figure 2. The first bone marrow evaluation was conducted at 3 years of age and revealed mildly hypocellular marrow (70% cellularity) with decreased and maturation-arrested granulopoiesis at the promyelocyte stage. Occasional large, atypical, binucleated, or multi-nucleated promyelocytes were reported. Using DNA harvested from peripheral blood, inherited neutropenia next-generation sequencing panel did not identify any mutations in genes known to be associated with SCN, including AP3B1, C16orf57, CSF3R, CXCR4, ELA2, G6PC3, GATA1, GATA2, GFI1, HAX1, LAMTOR2, LYST, RAB27A, RAC2, SDBS, SLC37A4, TAZ, VPS13B, VPS45, WAS, and WIPF1.
![Clinical features of the index patient. (A) Absolute neutrophil count (ANC) course since diagnosis. Clinically suspected pneumonia occurred at 6 years of age. Initially, the patient exhibited a clinical response with an increase in ANC with 5 µg/kg of G-CSF. However, there was no response in ANC rise within 14 days of a subsequent G-CSF treatment, even with dose escalation to 10 µg/kg. (B) The bone marrow (BM) aspirate smears exhibited granulocytic hypoplasia and promyelocytes/myelocytes with large size and multinucleation, and bizarre complex hypersegmentation in very large maturing granulocytes (red arrows) (Wright-Giesma stain, ×500). Results of the core biopsy showed cellular marrow with erythroid predominance, granulocytic hypoplasia, and small clusters of abnormal cells suspicious of left-shifted granulocytes with abnormal morphology seen in the aspirate smears (hematoxylin and eosin [H&E] stain, ×200). There is also increased apoptosis (green arrows). Immunohistochemistry (IHC) staining show granulocytic hypoplasia with a small number of cells stained with myeloperoxidase (black arrow). (C) The pedigree shows that only 1 of 3 children showed neutropenia and carried the LCP1 I232F mutation. The parents were asymptomatic and did not have the mutation in LCP1. The other 2 children were not affected and not tested for the mutation. (D) Schematic representation of LCP1 protein shows the domain organization and the location of the mutation within ABD1 in the CH1.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/8/10.1182_bloodadvances.2021006398/4/m_advancesadv2021006398f1.png?Expires=1764982122&Signature=YzJUQVkv-xznX0XJBrc6XtZXMLLpGbFNWnDttgxj9aTdvbOJnmYNut6miHweOCfm9raDOc51WUMBE7OiWaYgK2KHbsnZ1bfeUTHp0-ZtNrkVlOaoVghPEVDsRD4XhOUCmLA7Ds4CC0bgVeNa2Tmlf85RjeISKps~7VK2K25wcDtmGiIyl06UeInz6mx07do6O6y9r8nOTeCokUaV-TZoAW42GuCYiDIpPZdsrVs3YPDDR3sQRVW5CgLpgkterbQCpg38X0CnYu3icHYqBHhhK6SJq7lF2Jngof6BTHCAQNotaoO0PnEGsA18wivEX-qJIVOzyJT7LeXfFC8wG-6mUQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clinical features of the index patient. (A) Absolute neutrophil count (ANC) course since diagnosis. Clinically suspected pneumonia occurred at 6 years of age. Initially, the patient exhibited a clinical response with an increase in ANC with 5 µg/kg of G-CSF. However, there was no response in ANC rise within 14 days of a subsequent G-CSF treatment, even with dose escalation to 10 µg/kg. (B) The bone marrow (BM) aspirate smears exhibited granulocytic hypoplasia and promyelocytes/myelocytes with large size and multinucleation, and bizarre complex hypersegmentation in very large maturing granulocytes (red arrows) (Wright-Giesma stain, ×500). Results of the core biopsy showed cellular marrow with erythroid predominance, granulocytic hypoplasia, and small clusters of abnormal cells suspicious of left-shifted granulocytes with abnormal morphology seen in the aspirate smears (hematoxylin and eosin [H&E] stain, ×200). There is also increased apoptosis (green arrows). Immunohistochemistry (IHC) staining show granulocytic hypoplasia with a small number of cells stained with myeloperoxidase (black arrow). (C) The pedigree shows that only 1 of 3 children showed neutropenia and carried the LCP1 I232F mutation. The parents were asymptomatic and did not have the mutation in LCP1. The other 2 children were not affected and not tested for the mutation. (D) Schematic representation of LCP1 protein shows the domain organization and the location of the mutation within ABD1 in the CH1.
Clinical features of the index patient. (A) Absolute neutrophil count (ANC) course since diagnosis. Clinically suspected pneumonia occurred at 6 years of age. Initially, the patient exhibited a clinical response with an increase in ANC with 5 µg/kg of G-CSF. However, there was no response in ANC rise within 14 days of a subsequent G-CSF treatment, even with dose escalation to 10 µg/kg. (B) The bone marrow (BM) aspirate smears exhibited granulocytic hypoplasia and promyelocytes/myelocytes with large size and multinucleation, and bizarre complex hypersegmentation in very large maturing granulocytes (red arrows) (Wright-Giesma stain, ×500). Results of the core biopsy showed cellular marrow with erythroid predominance, granulocytic hypoplasia, and small clusters of abnormal cells suspicious of left-shifted granulocytes with abnormal morphology seen in the aspirate smears (hematoxylin and eosin [H&E] stain, ×200). There is also increased apoptosis (green arrows). Immunohistochemistry (IHC) staining show granulocytic hypoplasia with a small number of cells stained with myeloperoxidase (black arrow). (C) The pedigree shows that only 1 of 3 children showed neutropenia and carried the LCP1 I232F mutation. The parents were asymptomatic and did not have the mutation in LCP1. The other 2 children were not affected and not tested for the mutation. (D) Schematic representation of LCP1 protein shows the domain organization and the location of the mutation within ABD1 in the CH1.
During a hospitalization for facial cellulitis, fever, and neutropenia at age 4 years, the patient was treated with 2 daily doses of filgrastim (5 μg/kg per dose) (Figure 1A). During that hospitalization, a bone marrow biopsy revealed persistent hypocellularity (∼80%) with granulocytic hypoplasia, granulocytic maturation arrest at promyelocyte/myelocyte stage, and mildly increased myeloblasts (Figure 1B). In addition, there was significant granulocytic dysplasia with either a large size and/or hypersegmented nuclei in occasional maturing granulocytes and increased apoptosis. Because of the suspicion of myeloid neoplasia, an OnkoSight myeloid disorder sequencing panel (GenPath) was performed. No significant alternations were identified in genes associated with myeloid neoplasms. The patient also had mild normocytic anemia. Testing for Fanconi anemia (chromosomal breakage analysis with diepoxybutane and mitomycin C) and dyskeratosis congenita (telomere length) were within normal limits.
At the age of 4.5 years, the patient was again treated with a 6-day course of filgrastim during a hospitalization for fever and neutropenia. During this treatment, the patient did not respond to filgrastim therapy despite escalating the dose to 10 μg/kg (Figure 1A). A bone marrow biopsy was repeated; the results revealed normocellularity, a normal myeloid:erythroid ratio, granulocytic left-shift, and maturation to bands and segmented neutrophils with occasional binucleated forms. The granulocytic dysplasia observed in the prior examinations was improved. Eventually, in view of the unclear etiology of neutropenia and abnormal marrow morphology, whole-exome sequencing (GeneDx) was performed when the patient was 5 years of age. A heterozygous variant in the LCP1 gene (c.694 A>T in exon 7, which translates to LCP1 p.I232F) was detected by using a proprietary custom-developed analysis tool (XomeAnalyzer, GeneDx), by which data were filtered and analyzed to identify sequence variants and most deletions and duplications involving ≥3 coding exons.27 No variants in other genes associated with neutrophil disorders or neutropenia were reported. Family pedigree and the schematic structure of LCP1 are shown in Figure 1C-D. The LCP1 I232F is a novel variant not previously reported in the gnomAD or COSMIC databases.
To understand how I232F mutation of LCP1 could affect LCP1 binding to actin, we used a structural modeling approach to analyze the functional domains of LCP1. LCP1 contains EF-hand calcium-binding domains and 4 CH domains.28 In LCP1, two tandem CH domains (CH1-CH2 and CH3-CH4) formed 2 actin-binding domains (ABD1 and ABD2) that bind to actin.28-30 No ABD1 of the LCP1 structure is reported, but the ABD2 of LCP1 (LCP1-ABD2) with F-actin structure is known (Figure 2A) (Protein Data Bank identifier, 6VEC).30 The complex structure of LCP1-ABD2 with F-actin showed that only CH3 of LCP1-ABD2 interacts with F-actin, and CH3 and CH4 have a similar 4-core helices fold31 that are connected by an interdomain helix (Figure 2A,D). Of note, the structure between LCP1-ABD2 and F-actin30 is similar to the complex structure of actin and fimbrin ABD2,29 implying a conserved mode of interaction between a tandem CH domain with actin.
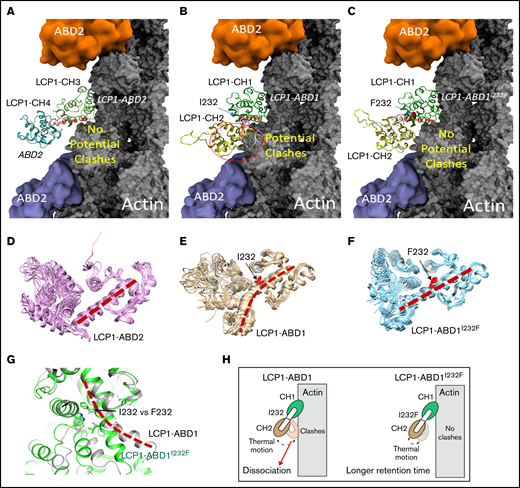
Structural modeling of the LCP1 I232F mutation. (A) Cryo-EM structure of LCP1-ABD2 (Protein Data Bank identifier, 6VEC). Final snapshots of the molecular dynamics–generated LCP1-ABD1 (B) and LCP1-ABD1I232F (C) structure aligned to LCP1-ABD2 to model the binding between LCP1-ABD1 and F-actin. Ten representative conformations of LCP1-ABD2 (D), LCP1-ABD1 (E), and LCP1-ABD1I232F (F) obtained from 40 ns of molecular dynamics simulations are shown to depict the dynamic motion of the protein. The interdomain helices between CH1 and CH2 in LCP1-ABD1 and between CH3 and CH4 in LCP1-ABD2 are highlighted by red dashed lines. I232 and F232 are shown in red stick models. Potential clashes between LCP1-ABD1 and F-actin is indicated in a red circle. (G) Alignment of LCP1-ABD1-CH1 and LCP1-ABD1I232F-CH1. The bent interdomain helix in LCP1-ABD1 is depicted in a red dashed line. (H) A model illustrates the effect of I232F mutation to LCP1-ABD1 on the binding of LCP1-ABD1 to F-actin.
Structural modeling of the LCP1 I232F mutation. (A) Cryo-EM structure of LCP1-ABD2 (Protein Data Bank identifier, 6VEC). Final snapshots of the molecular dynamics–generated LCP1-ABD1 (B) and LCP1-ABD1I232F (C) structure aligned to LCP1-ABD2 to model the binding between LCP1-ABD1 and F-actin. Ten representative conformations of LCP1-ABD2 (D), LCP1-ABD1 (E), and LCP1-ABD1I232F (F) obtained from 40 ns of molecular dynamics simulations are shown to depict the dynamic motion of the protein. The interdomain helices between CH1 and CH2 in LCP1-ABD1 and between CH3 and CH4 in LCP1-ABD2 are highlighted by red dashed lines. I232 and F232 are shown in red stick models. Potential clashes between LCP1-ABD1 and F-actin is indicated in a red circle. (G) Alignment of LCP1-ABD1-CH1 and LCP1-ABD1I232F-CH1. The bent interdomain helix in LCP1-ABD1 is depicted in a red dashed line. (H) A model illustrates the effect of I232F mutation to LCP1-ABD1 on the binding of LCP1-ABD1 to F-actin.
Because no LCP1-ABD1 structure is available, the I-TASSER program17,18 was used to obtain 5 models of LCP1-ABD1 (sequence, 120-380) (Figure 2B,E) through homology modeling. All models of LCP1-ABD1 exhibited a similar protein fold as LCP1-ABD2, and we selected the first and the highest scored model to represent LCP1-ABD1. The backbone root-mean-squared deviations between the LCP1-ABD1 model and LCP1-ABD2 from the cryo-EM structure were 3.30 Å, which was attributed to variation of the loop regions and a short helix-loop between E127 and P147 in LCP1-ABD1. Based on the LCP1-ABD1 model, the I232F mutation is located at the interdomain helix between CH1 and CH2 in LCP1-ABD1 (Figure 2B-C, E-F). After alignment of LCP1-ABD1 and LCP1-ABD2, we hypothesized that CH1 in LCP1-ABD1 binds to F-actin similarly to CH3 in LCP1-ADB2 (Figure 2A-B). In this model, I232F in LCP1-ABD1 is not expected to affect direct interactions between LCP1-CH1 and F-actin binding directly.
We then questioned if the I232F mutation may affect the dynamical motion of LCP1-ABD1 and have an allosteric influence on interactions between LCP1-ABD1 and F-actin. To model the thermal motion of the proteins, we performed 40 ns of molecular dynamics simulations of LCP1-ABD1, LCP1-ABD1I232F, and LCP1-ABD2 (Figure 2D-F). Based on the simulations of LCP1-ABD2, the interdomain helix retained rigidity that limited the motion of LCP1-CH4 (Figure 2D). For LCP1-ABD1, the interdomain helix exhibited a higher degree of flexibility and adopted a bent helix conformation (Figure 2E). In contrast, the interdomain helix in LCP1-ABD1I232F gained rigidity and showed similar dynamic motion as LCP1-ABD2 (Figure 2F). After aligning the final snapshot of LCP1-ABD1 and LCP1-ABD1I232F to LCP1-ABD2 in the LCP1-ABD2/F-actin structure, we found that the bent helix in LCP1-ABD1 may cause a close contact of CH2 with F-actin when LCP1-ABD1 underwent dynamical motion (Figure 2G). In comparison, the rigid interdomain helix, resulting from I232F mutation, in LCP1-ABD1 positioned CH2 to avoid potential clashes with F-actin similarly to CH4 in LCP1-ABD2. Based on our model, we proposed that I232F in LCP1-ABD1 caused LCP1-ABD1I232F to adopt a relatively rigid conformation and reduced the potential unfavorable clashes between CH2 in LCP1-ABD1 and F-actin over time. The consequence is that LCP1-ABD1I232F remains associated with F-actin at a longer period of time than LCP1-ABD1 with F-actin (Figure 2H). The proposed mechanism differs from the mutations found in plastin 3 in which mutations occur either at the actin-binding loop to disrupt plastin 3–actin binding or in other regions to decrease Ca2+ sensitivity.30
LCP1 I232F impairs granulocytic proliferation in 32D cells secondary to cell cycle arrest
To investigate the effect of LCP1 I232F on granulocytic proliferation, we used doxycycline-inducible expression of wtLCP1 and LCP1 I232F in 32D cells. The 32D cells are murine myeloblast cells, which are dependent on IL-3 for their growth, survival, and proliferation. In addition, they differentiate to neutrophils upon treatment with G-CSF. In our model, the induction of wtLCP1 and LCP1 I232F occurred with 1 μg/mL of doxycycline at both the protein and gene expression level (supplemental Figure 1). The LCP1 I232F-expressing 32D cells exhibited decreased proliferation with no impairment of cell viability (Figure 3A). We also observed the appearance of large dysplastic cells with multilobulated nuclei and nuclear-cytoplasmic asynchrony with the overexpression of LCP1 I232F (Figure 3B). Furthermore, cell cycle analysis showed cell cycle arrest at the G2/M phase in LCP I232F-expressing cells, suggesting failure of cytokinesis (Figure 3C). This was accompanied by upregulation of markers of cell cycle arrest such as p21 (Cdkn1a), Cdkn1b, and Tp53, but they did not reach statistical significance (supplemental Figure 3).
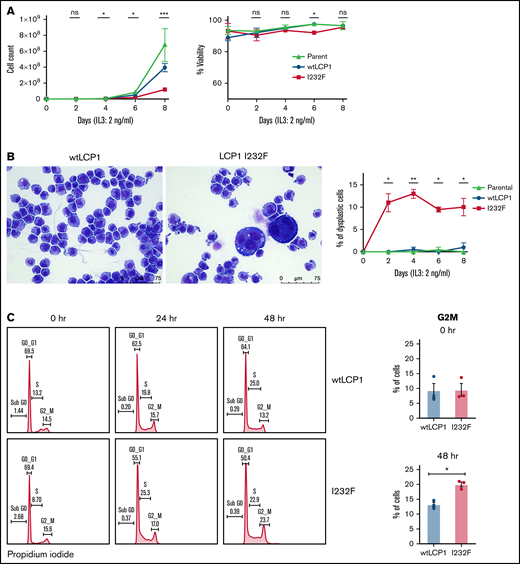
Effect of overexpression of LCP1 I232F in 32D cell proliferation. (A) 32D cells expressing LCP1 I232F display impaired cell proliferation with unchanged viability upon proliferation with murine IL-3–containing media (2 ng/mL). (B) Images, obtained at ×40 magnification using a Leica DM6B upright microscope, show that LCP1 I232F-expressing cells have granulocytic dysplasia with the appearance of large multinucleated cells. (C) The cell cycle analysis for 32D cells harvested at indicated time points using DNA staining with propidium iodide followed by flow cytometric analysis (left) shows arrest of cell cycle at G2M Phase in LCP1 I232F-expressing cells (right). Data are shown as mean ± standard error of mean (results of n = 3 biological replicates unless otherwise specified). Statistical analysis was assessed by Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
Effect of overexpression of LCP1 I232F in 32D cell proliferation. (A) 32D cells expressing LCP1 I232F display impaired cell proliferation with unchanged viability upon proliferation with murine IL-3–containing media (2 ng/mL). (B) Images, obtained at ×40 magnification using a Leica DM6B upright microscope, show that LCP1 I232F-expressing cells have granulocytic dysplasia with the appearance of large multinucleated cells. (C) The cell cycle analysis for 32D cells harvested at indicated time points using DNA staining with propidium iodide followed by flow cytometric analysis (left) shows arrest of cell cycle at G2M Phase in LCP1 I232F-expressing cells (right). Data are shown as mean ± standard error of mean (results of n = 3 biological replicates unless otherwise specified). Statistical analysis was assessed by Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
LCP1 I232F does not increase apoptosis or unfolded protein response
To investigate whether LCP1 I232F leads to increased cell death as observed in the index patient, we stained 32D cells growing in IL-3 media with Annexin V and propidium iodide. 32D LCP1 I232F cells did not show increased apoptosis according to flow cytometry (Figure 4A). 32D cells were additionally challenged with IL-3 withdrawal, leading to increased cell death. However, stress induction in 32D cells elicited increased apoptosis upon LCP1 I232F expression (Figure 4B). We further investigated the markers of apoptosis by immunoblotting, which did not reveal increased evidence of apoptosis (Figure 4C). To identify other sources of intracellular stress, we examined markers of unfolded protein response (UPR) but observed no increased UPR in LCP1 I232F-expressing cells (Figure 4D).

Effect of LCP1 I232F on cellular stress responses. (A) Flow cytometric analysis of Annexin V and propidium iodide (PI)-based apoptosis assay was performed on isogenic 32D cell lines as indicated. Camptothecin (CPT; 1 μM) was used as positive control. (B) 32D cells (wtLCP1 and LCP1 I232F) grown under conditions of IL-3 withdrawal for 48 hours were analyzed for apoptosis by flow cytometry–based Annexin V/PI staining (n = 3). Annexin V+ and Annexin V+/PI+ cells were identified as early and late apoptotic cells, respectively. (C) Analysis apoptotic markers, PARP1 and Caspase-3, for their cleavage by immunoblotting was performed on 32D cells expressing wtLCP1 and LCP1 I232F as indicated. β-actin was used as loading control. (D) Real-time quantitative polymerase chain reaction analysis investigating makers of unfolded protein response Bip, Chop, Xbp1/Xbp1s, and Atf4 was performed at indicated time points with Actin as the housekeeping gene (n = 4). Data are shown as mean ± standard deviation. Statistical analysis was assessed by using the Student t test. dox, doxycycline; DMSO, dimethyl sulfoxide.
Effect of LCP1 I232F on cellular stress responses. (A) Flow cytometric analysis of Annexin V and propidium iodide (PI)-based apoptosis assay was performed on isogenic 32D cell lines as indicated. Camptothecin (CPT; 1 μM) was used as positive control. (B) 32D cells (wtLCP1 and LCP1 I232F) grown under conditions of IL-3 withdrawal for 48 hours were analyzed for apoptosis by flow cytometry–based Annexin V/PI staining (n = 3). Annexin V+ and Annexin V+/PI+ cells were identified as early and late apoptotic cells, respectively. (C) Analysis apoptotic markers, PARP1 and Caspase-3, for their cleavage by immunoblotting was performed on 32D cells expressing wtLCP1 and LCP1 I232F as indicated. β-actin was used as loading control. (D) Real-time quantitative polymerase chain reaction analysis investigating makers of unfolded protein response Bip, Chop, Xbp1/Xbp1s, and Atf4 was performed at indicated time points with Actin as the housekeeping gene (n = 4). Data are shown as mean ± standard deviation. Statistical analysis was assessed by using the Student t test. dox, doxycycline; DMSO, dimethyl sulfoxide.
LCP1 I232F produces dysplastic granulocytes in vitro
Many of the SCNs occur secondary to a block in differentiation at the promyelocyte/myelocyte stage. To investigate the role of LCP1 I232F in granulocytic differentiation, 32D cells were treated with G-CSF (100 ng/mL) for 8 days. LCP1 I232F did not affect cell count or viability (Figure 5A). However, LCP1 I232F-expressing 32D cells displayed perturbed granulocytic differentiation with an increased number of dysplastic cells, as seen on IL-3–induced proliferation (Figure 5B). Furthermore, the flow cytometric evaluation using CD11b and Gr-1 antibodies exhibited somewhat decreased staining of mutant LCP1 I232F cells with CD11b (Figure 5C) but did not display a significant block in differentiation (supplemental Figure 4). Although master transcription factors Gfi1 and Spi1 were decreased on day 2 of 32D cell differentiation by G-CSF, the levels recovered thereafter. Terminal differentiation markers and transcription factors did not exhibit significant differences except Cebpe on day 6 (Figure 5D). Interestingly, we observed small but statistically significant upregulation of CXCR4, the gene that encodes the receptor for CXCL12/SDF-1. (A truncating mutation in CXCR4 causes WHIM syndrome characterized by warts, hypogammaglobinemia, infections, and myelokathexis leading to neutropenia).
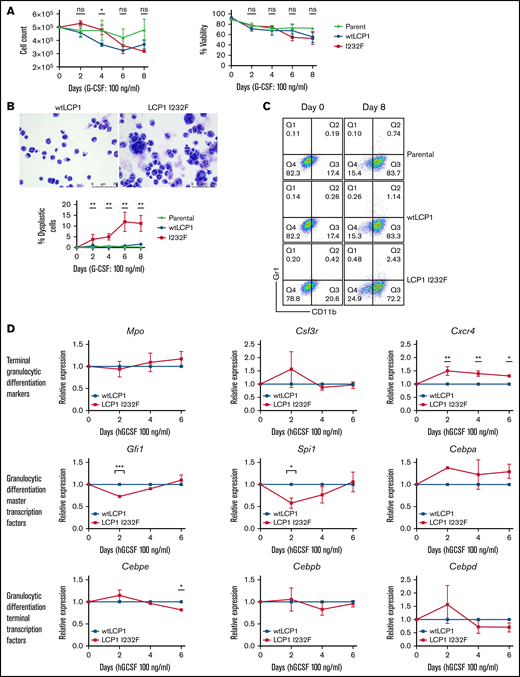
Effect of LCP1 I232F on G-CSF–induced granulocytic differentiation. (A) Trypan blue exclusion cell count and viability were performed during differentiation of LCP1 I232F-expressing 32D cells with G-CSF treatment (100 ng/µL) for 8 days. (B) Wright-Giemsa–stained images, obtained by using a Leica DM6B upright microscope (×40 magnification), for 32D cells expressing wtLCP1 and LCP1 I232F upon differentiation with G-CSF (left and middle) and analysis of large dysplastic cells (right). (C) LCP1 (wt and I232F) expressing 32D cells differentiated with G-CSF were stained with anti-CD11b and anti–GR-1 antibodies and analyzed with flow cytometry. (D) 32D cells expressing wtLCP1 and LCP1 I232F were differentiated with G-CSF followed by quantitative polymerase chain reaction–based gene expression analysis of indicated master transcription factors of granulopoiesis and terminal granulocytic differentiation markers and transcription factors at days 0, 2, 4, and 6. Data are shown as mean ± standard error of mean (results of n = 3 biological replicates unless otherwise specified). Statistical analysis was assessed by using the Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. hGCSF, human G-CSF; ns, not significant.
Effect of LCP1 I232F on G-CSF–induced granulocytic differentiation. (A) Trypan blue exclusion cell count and viability were performed during differentiation of LCP1 I232F-expressing 32D cells with G-CSF treatment (100 ng/µL) for 8 days. (B) Wright-Giemsa–stained images, obtained by using a Leica DM6B upright microscope (×40 magnification), for 32D cells expressing wtLCP1 and LCP1 I232F upon differentiation with G-CSF (left and middle) and analysis of large dysplastic cells (right). (C) LCP1 (wt and I232F) expressing 32D cells differentiated with G-CSF were stained with anti-CD11b and anti–GR-1 antibodies and analyzed with flow cytometry. (D) 32D cells expressing wtLCP1 and LCP1 I232F were differentiated with G-CSF followed by quantitative polymerase chain reaction–based gene expression analysis of indicated master transcription factors of granulopoiesis and terminal granulocytic differentiation markers and transcription factors at days 0, 2, 4, and 6. Data are shown as mean ± standard error of mean (results of n = 3 biological replicates unless otherwise specified). Statistical analysis was assessed by using the Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. hGCSF, human G-CSF; ns, not significant.
LCP1 I232F impairs cell migration and invasiveness in vitro but does not impair oxidative burst
The cytoskeleton provides the structural scaffold for the cellular architecture and stability. Actin and various actin-binding proteins along with microtubules and intermediate filaments are involved in essential cellular processes such as migration, organelle and vesicle transport, phagocytosis, and axonal growth. LCP1 is an important actin-binding protein. Its role in cell proliferation and metastasis has been described in various cancers.32-36 To investigate how LCP1 I232F affects cell migration, we performed a scratch wound-healing assay using HeLa cells. We observed that the wound closure was impaired in LCP1 I232F-expressing cell monolayers (Figure 6A-B). We further observed decreased migratory movement of 32D and HeLa cells in Boyden chamber assay (Figure 6C-D). Interestingly, compared with the parental and wtLCP1 cells, the 32D cells expressing LCP1 I232F exhibited similar reduced NAD phosphate–dependent reactive oxygen species formation upon activation with N-formyl-Met-Leu-Phe and phorbol myristate acetate (Figure 6E).
![Effect of LCP1 I232F on cell migration and invasiveness. HeLa cells expressing wtLCP1 and LCP1 I232F were examined by using a scratch wound–healing assay at indicated time points (A), and results were analyzed graphically (n = 5) (B). Boyden chamber cell migration of HeLa (C) and 32D (D) cells (above) expressing wtLCP1 and LCP1 I232F. The assay was quantified by cell count and optical density (below). (E) Flow cytometry for the analysis of oxidative burst of indicated 32D cells, which were differentiated with 8 days of G-CSF treatment followed by stimulation with N-formyl-Met-Leu-Phe (fMLP) and phorbol myristate acetate (PMA). Oxidative burst was measured after treatment with dihydrorhodamine-123 (reported as mean fluorescence intensity [MFI] normalized to DFMO-treated samples). *P < .05, **P < .01, ***P < .001, ****P < .0001. DMSO, dimethyl sulfoxide; ns, not significant; PE, phycoerythrin; PE-A, Phycoerythrin-A.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/8/10.1182_bloodadvances.2021006398/4/m_advancesadv2021006398f6.png?Expires=1764982122&Signature=3ecZa3Al4um4fXL07qEpWvF11~PHTQWhN2Bn9m6PNq2AYPeV8DZJo9o~FMJ~VgiaksyOb6z56CmqYnpr5lCSIPZ--HBvdIuPGvvbcqo-dqBRce6OkiZGTxsXyIIpyy0d3w~BkEmnKDNlP8axo~MffQhcRlilmYG5JLHBFRSFM5TuBCybAuQ3EUY2tuv9EbjATaHYuQbikuMtk4VcYtK9FoxjoiO0v7CPCOjkuSP3QtgfhmxoNd5SMpgAACEg-zbxslPmNwIl5dApS4rzuKs2wJo2FZZC1eACpnS0ZCG6Ilre3Z72HH4HMCc7dSxO7ljLRJrgoAaz1BiLwhpF1LIjRA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of LCP1 I232F on cell migration and invasiveness. HeLa cells expressing wtLCP1 and LCP1 I232F were examined by using a scratch wound–healing assay at indicated time points (A), and results were analyzed graphically (n = 5) (B). Boyden chamber cell migration of HeLa (C) and 32D (D) cells (above) expressing wtLCP1 and LCP1 I232F. The assay was quantified by cell count and optical density (below). (E) Flow cytometry for the analysis of oxidative burst of indicated 32D cells, which were differentiated with 8 days of G-CSF treatment followed by stimulation with N-formyl-Met-Leu-Phe (fMLP) and phorbol myristate acetate (PMA). Oxidative burst was measured after treatment with dihydrorhodamine-123 (reported as mean fluorescence intensity [MFI] normalized to DFMO-treated samples). *P < .05, **P < .01, ***P < .001, ****P < .0001. DMSO, dimethyl sulfoxide; ns, not significant; PE, phycoerythrin; PE-A, Phycoerythrin-A.
Effect of LCP1 I232F on cell migration and invasiveness. HeLa cells expressing wtLCP1 and LCP1 I232F were examined by using a scratch wound–healing assay at indicated time points (A), and results were analyzed graphically (n = 5) (B). Boyden chamber cell migration of HeLa (C) and 32D (D) cells (above) expressing wtLCP1 and LCP1 I232F. The assay was quantified by cell count and optical density (below). (E) Flow cytometry for the analysis of oxidative burst of indicated 32D cells, which were differentiated with 8 days of G-CSF treatment followed by stimulation with N-formyl-Met-Leu-Phe (fMLP) and phorbol myristate acetate (PMA). Oxidative burst was measured after treatment with dihydrorhodamine-123 (reported as mean fluorescence intensity [MFI] normalized to DFMO-treated samples). *P < .05, **P < .01, ***P < .001, ****P < .0001. DMSO, dimethyl sulfoxide; ns, not significant; PE, phycoerythrin; PE-A, Phycoerythrin-A.
LCP1 I232F increases F-actin polymerization in vitro and causes mislocalization of LCP1 to the nucleus
LCP1 binds to F-actin filaments to cross-link or stabilize parallel strands of actin filaments.28 X-linked SCN due to gain-of-function WAS mutation results from enhanced and delocalized F-actin polymerization, decreased proliferation, and increased apoptosis.12 To investigate how LCP1 I232F overexpression affects actin polymerization, we analyzed LCP1 or LCP1 I232F-expressing cells by confocal microscopy after dual fluorescence labeling with anti-LCP1 antibody and an F-actin probe, phalloidin. The mutant-expressing HeLa cells exhibited increased levels of F-actin and enrichment of F-actin and LCP1 at the cell cortex (Figure 7A,C). The flow cytometric evaluation of LCP1 I232F-mutant 32D cells confirmed the presence of increased levels of intracellular F-actin, as previously reported in WASp-associated SCN (Figure 7B). Interestingly, mutant LCP1-expressing HeLa cells exhibited irregular nuclear contour with folds and indentations, and the mislocalization of LCP1 inside the nucleus, which was further confirmed with subcellular fractionation (Figure 7C-D).

Effect of LCP1 I232F overexpression on actin polymerization. (A) 32D cells expressing LCP1 I232F with dysplastic features were dual fluorescently labeled with anti-LCP1 antibody (red color) and an F-actin probe, phalloidin (green color). The labeled cells were imaged by confocal microscopy. The mutant-expressing cells exhibit increased level of F-actin and enrichment of F-actin and LCP1 at the cell cortex. (B) Flow cytometric quantification of F-actin levels in 32D cells expressing either wtLCP1 or I232F mutant grown under indicated conditions. (C) HeLa cells expressing either wtLCP1 or I232F mutant were imaged by confocal microscopy after dual labeling with anti-LCP1 antibody and phalloidin. (D) Subcellular fractionation of 32D cells expressing LCP1 (wt and I232F) upon 3 days of IL-3 treatment. DAPI, 4′,6-diamidino-2-phenylindole; H2AX, H2A histone family member X; MEK, mitogen activated protein kinase kinase; PARP1, poly (ADP-ribose) polymerase 1; VDAC, voltage-dependent anion channel.
Effect of LCP1 I232F overexpression on actin polymerization. (A) 32D cells expressing LCP1 I232F with dysplastic features were dual fluorescently labeled with anti-LCP1 antibody (red color) and an F-actin probe, phalloidin (green color). The labeled cells were imaged by confocal microscopy. The mutant-expressing cells exhibit increased level of F-actin and enrichment of F-actin and LCP1 at the cell cortex. (B) Flow cytometric quantification of F-actin levels in 32D cells expressing either wtLCP1 or I232F mutant grown under indicated conditions. (C) HeLa cells expressing either wtLCP1 or I232F mutant were imaged by confocal microscopy after dual labeling with anti-LCP1 antibody and phalloidin. (D) Subcellular fractionation of 32D cells expressing LCP1 (wt and I232F) upon 3 days of IL-3 treatment. DAPI, 4′,6-diamidino-2-phenylindole; H2AX, H2A histone family member X; MEK, mitogen activated protein kinase kinase; PARP1, poly (ADP-ribose) polymerase 1; VDAC, voltage-dependent anion channel.
Discussion
We report a novel cause of symptomatic neutropenia associated with a missense mutation LCP1 c.694 A>T (p.I232F), a gene encoding an F-actin–binding protein. To date, no variants of LCP1 have been reported with any human disease. The variant identified in our patient occurs in the first CH domain (ie, CH1), which binds to F-actin. The actin-binding domains of accessory actin-binding proteins are known to be sensitive to actin filament conformational changes.37 However, how CH1 domain mutations of LCP1 alter its actin bundling property has not been established. Our structural modeling shows that LCP1-ABD1I232F remains associated with F-actin for a longer period of time than LCP1-ABD1. This could explain the increased amount of F-actin exhibited in our cell models. Our model suggests that I232F rigidifies LCP1 I232F (relative to wtLCP1) to reduce clashes between LCP1-CH2 and actin due to thermal motion, thereby prolonging the binding between LCP1 I232F and actin. In the heterozygous condition, wtLCP1 will compete with LCP1 I232F on actin binding, and the cellular phenotype will depend on the relative concentrations of the wt and mutant LCP1. This argues for our observation of a consistent 10% large dysmorphic granulocyte population in cells expressing the mutant, which phenocopied the patient’s dysplastic neutrophils. Furthermore, the mutant form inhibited proliferation with a G2/M cell cycle arrest, but it did not induce apoptosis, TP53 activation, or UPR. The mutant form, but not the wt form, mislocalized to the nucleus. Our results suggest that nuclear mislocalization of mutant LCP1 affects the number of circulating neutrophil by inducing a G2/M arrest, dysplasia, and impaired proliferation.
LCP1 is one of the plastins, which are highly conserved. In mammals, there are 3 plastin isoforms: plastin 1 (I-plastin) is expressed only in renal and intestinal epithelium, plastin 2 (L-plastin, LPL, or LCP1) is expressed only in hematopoietic cells, and plastin 3 (T-plastin) is broadly expressed.38 LCP1 plays an important role in leukocyte biology by crosslinking F-actin filaments, thereby contributing to cytoskeletal stability for critical signaling pathways.39 LCP1 functions as an actin-bundling protein and enhances dynamic architecture of neutrophil cytoskeleton important in neutrophil migration and function. In addition, it associates with the intermediate filament protein vimentin at adhesion sites. Animal studies have shown that the LCP1-deficient neutrophils are deficient in killing Staphylococcus aureus, despite normal phagocytosis.40 Loss of LCP1 has been shown to impair T-cell polarization, migration, and T-cell–dependent antibody responses.38,41,42 Kell et al28 generated lcp1−/− zebrafish using CRISPR-Cas9 editing. They observed that lcp1−/− fish develop and reproduce normally, but they display reduced survival, with mortality ∼4 to 6 weeks’ postfertilization. Our studies suggest that the I232F mutation in LCP1 is not the same as a loss of LCP1 and that the LCP1 mutation could promote F-actin bundling. Studies with knockout animal models did not observe neutropenia, nor did they report large dysplastic cells, which further support a role for the mutation I232F in neutropenia.
Our findings suggest that expression of LCP1 I232F caused granulocytic dysplasia and impaired cell proliferation and motility leading to neutropenia. Such dysplasia is represented by large dysmorphic granulocytes forming ∼10% of the cell population, which reiterates cellular heterogeneity due to cell cycle distribution, transcriptional noise, or stochasticity. The neutropenia may be due to increased F-actin polymerization that underlies the cell cycle arrest and nuclear mislocalization. Our patient’s phenotype closely resembles WASp X-linked neutropenia. We propose that with the identification of autosomal dominant mutation of LCP1, cytoskeletal dysregulation explains many forms of the inherited neutropenias. These include mutations of CXCR4 (receptor for the chemokine CXCL12); DNM2 (mechano-GTPase);43 WAS (nucleator for actin polymerization); BTK (kinase that interacts with WASp);44 HAX1, which associates with HCLS1 (actin-binding protein);45 WDR1 (actin-binding protein);46 VPS45 and VPS13B (vacuolar protein sorting mediators);7,47 and MKL1 (transcription factor affecting cytoskeleton-associated genes).48 Hence, actin dynamics may be as critical for production of circulating neutrophils as they are for neutrophil functions.
Acknowledgments
The authors acknowledge the index patient and her family.
This work was supported in part by grants from the National Institutes of Health R01 128173 (S.J.C.), DOD Idea Award (S.J.C.), and VeloSano (S.J.C., H.M.).
Authorship
Contribution: U.M., B.G., and H.M. performed the experiments and analyzed data; C.-Y.Y. performed the modeling; U.M., B.G., and S.J.C. conceptualized the study and designed the experiments; A.I.I. provided guidance with actin cytoskeletal immunofluorescence staining and visualization; H.J.R. reviewed the bone marrow examinations of the index patient and provided images; R.H. and A.F. provided relevant clinical information of the index patient; and U.M., C.-Y.Y., and S.J.C. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Seth J. Corey, Department of Pediatric Hematology/Oncology and SCT, 9500 Euclid Ave, Mail R-3, Cleveland, OH 44195; e-mail: coreylab@yahoo.com.

