

TO THE EDITOR:
The World Health Organization and International Consensus Classification classifications of myeloid neoplasms define a category of myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions.1-3 The most frequent neoplasms in this category are associated with FIP1L1::PDGFRA (F/P) fusions, created because of a somatic, acquired interstitial deletion on chromosome 4q12.4-7 Patients with F/P or rearrangements of PDGFRB may present with hypereosinophilic syndrome (HES), defined as >1.5 × 106 eosinophils/mL blood and evidence of eosinophil-mediated end–organ manifestations.8,9 Patients with PDGFR-rearranged HESN (HES related to a primary myeloid neoplasm) are mostly males, and the hypereosinophilia responds robustly to the multikinase inhibitor imatinib, which targets the constitutive PDGF receptor signaling driven by the fusions.3-7 Our goal was to investigate HESN whole genomes to better characterize the disease and its unique features.
Unbiased genomics of PDGFR-rearranged HES have not been comprehensively studied at genome scale, with only small, targeted sequencing panel data reported.10 We performed whole-genome sequencing (WGS) on purified eosinophils (tumor) from 11 patients with chronic phase PDGFR–related HESN (supplemental Table 1; supplemental Methods). Paired normal samples were peripheral blood mononuclear cells collected in hematologic and molecular remission after imatinib treatment, in the setting of normal eosinophil counts and no detectable PDGFR rearrangement (Figure 1A; supplemental Figure 1A). The median age at tumor sampling was 43 years (range, 13-60; supplemental Table 1); there were 10 males and 1 female (expected sex bias5). Nine males had evidence of an F/P fusion. The female had a PDGFRB::ETV6 rearrangement, and 1 male had a novel complex rearrangement involving PDGFRB (as described later in the article). The mean sequencing depth was 83 (range, 61-144) for tumor and 91 (range, 67-125) for normal (supplemental Figure 1B; supplemental Table 2).

Genomic characterization of PDGFR rearrangements. (A) Absolute eosinophil count before (tumor) and after treatment (normal) with imatinib. (B) GISTIC2 plot depicting the significantly recurrent interstitial deletion on chromosome 4. (C) Genomic view of the F/P locus showing detected deletions (blue bars) in most samples. (D) Detailed view of the breakpoints inside the FIP1L1 gene. Colored lines show location inside introns labeled with matching intron numbers (based on transcript NM_001134937.1). (E) Circos plot11 of the complex PDGFRB rearrangement in patient HES11. Purple lines indicate genomic fusions between the chromosomal regions. (F) Gene locus and inferred protein fusion between PDGFRB and IQGAP2.
Genomic characterization of PDGFR rearrangements. (A) Absolute eosinophil count before (tumor) and after treatment (normal) with imatinib. (B) GISTIC2 plot depicting the significantly recurrent interstitial deletion on chromosome 4. (C) Genomic view of the F/P locus showing detected deletions (blue bars) in most samples. (D) Detailed view of the breakpoints inside the FIP1L1 gene. Colored lines show location inside introns labeled with matching intron numbers (based on transcript NM_001134937.1). (E) Circos plot11 of the complex PDGFRB rearrangement in patient HES11. Purple lines indicate genomic fusions between the chromosomal regions. (F) Gene locus and inferred protein fusion between PDGFRB and IQGAP2.
Few segmental DNA copy number alterations were detected, and the interstitial deletion between FIP1L1 and PDGFRA was the only recurrent event (Figure 1B). F/P was also the most frequently identified structural variant (SV) (supplemental Table 1). No evidence of F/P was detected in paired normal samples. All breakpoints in PDGFRA occurred within exon 12 (Figure 1C). The FIP1L1 breakpoints spanned a region of ∼45 kb covering introns 8 to 12 (Figure 1D; supplemental Table 1).
SV analysis confirmed PDGFRB fusions in 2 tumors with PDGFRB events using fluorescence in situ hybridization (supplemental Table 1). One was a classic ETV6::PDGFRB fusion.12 The other comprises a previously unreported rearrangement involving IQGAP2 and PDGFRB on chromosome 5 and UVRAG on chromosome 11. This SV created an in-frame protein fusion between exon 10 of IQGAP2 and exon 12 of PDGFRB, with the other ends of these breakpoints connecting to the first intron of UVRAG (Figure 1E-F; supplemental Figure 1C).
Single-nucleotide variants (SNVs) and insertions and deletions occurred at a median rate of 0.37 events per megabase (range, 0.23-0.58), comparable with that of myeloproliferative neoplasms (0.38 mutations per megabase13) and lower than that of other hematologic malignancies (Figure 2A). The absolute number of SNVs and insertions and deletions in HESN tumors ranged from 653 to 1652 (Figure 2B; supplemental Figure 2A).
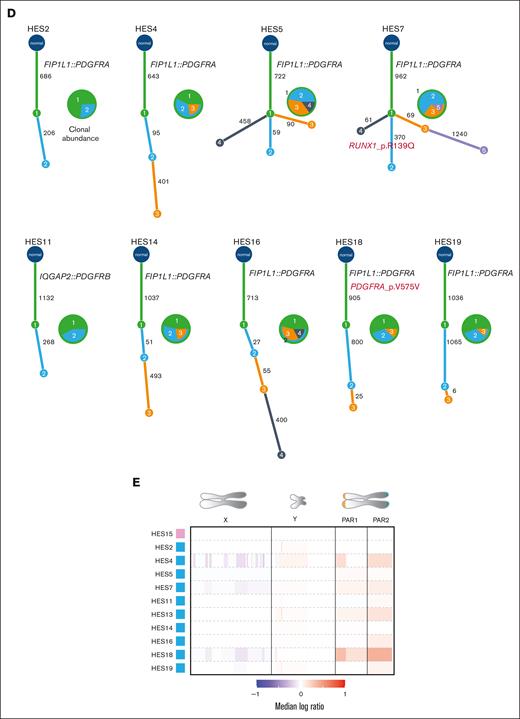
Genomic analyses of PDGFR-rearranged HESN. (A) Mutation rate comparison between HESN and other myeloid malignancies. Each red dot indicates a sample. (B) Number of SNVs (blue), deletions (orange), and insertions (green). (C) Mutational signature profile for HES samples. The height of each bar indicates the number of mutations in each context (on the x-axis) and base change type (distinguished by color). (D) Inferred phylogenies for single tumors. Pie charts show the abundance of each clone in each sample. Numbers inside the pie segments correspond to clone ID numbers on the tree. Numbers on tree branches indicate the number of mutations in each branch. Protein-coding alterations in known cancer genes are marked in red. (E) Copy number calls as median log ratio (tumor:normal) for chrX and chrY and 2 pseudoautosomal regions. Pink box: female; blue box: male. AML, acute myeloid leukemia; BNHL, B-cell non-Hodgkin lymphoma;13 CLL, chronic lymphocytic leukemia; MPN, myeloid proliferative neoplasm.

Signature analysis revealed COSMIC SBS5 (a DNA mutation pattern enriched for C>T transitions14,15) as the most frequent tumor-associated abnormality (cosine similarity, 0.77) (Figure 2C; supplemental Figure 2). SBS5 is found across all tumor types, including myeloid neoplasms, and exhibits clock-like behavior that correlates with age. No signature suggesting exposure to environmental (eg, cigarette smoke or ultraviolet light) or endogenous (eg, activation-induced cytidine deaminase [AID]) mutagens was detected.
Next, we investigated clonal structure using single-sample inference.16 Most tumors exhibited linear phylogenies with 1 or 2 subclones (Figure 2D), with 2 (HES5 and HES7) having branched evolution. The PDGFR fusion occurred in the truncal clone in all cases. We detected a subclonal missense mutation in RUNX1 in HES7 causing an Arg139Gln amino acid change, previously observed in myeloid disorders.17,18 In total, 87 nonsilent somatic mutations in protein-coding genes were identified in HESN tumors, with no recurrently mutated genes. With the caveat of small cohort size, this suggests that there are no obvious additional somatic driver mutations in HESN beyond the PDGFR rearrangement.
Identifying recurrent somatic, noncoding driver mutations is challenging because they may not involve the identical base, and there is no exonic structure to guide variant classification.19 Our WGS depth provided sufficient power to interrogate GC-rich promoters and 5′ untranslated regions (UTRs), which are underrepresented in older studies (supplemental Figure 3).19 To focus on potential eosinophil-relevant noncoding regions, we analyzed H3K27 acetylation (active promoters and enhancers) and CCCTC-binding factor (CTCF, a marker of chromatin regulatory hub boundaries) sites from chromatin immunoprecipitation sequencing in EOL1 eosinophilic leukemia cells, which harbor a F/P fusion. No recurrent mutations (>1 mutation per element) were found in these regions or in transcription start sites, 3′UTRs, and 5′UTRs of protein-coding genes.
Given that the paired normal samples were peripheral blood mononuclear cells collected from patients during hematologic and molecular remission, we searched for background clonal hematopoiesis (CH) in 159 CH-associated genes.20 Only 1 nonsilent variant was identified, a CSF1R substitution (p.R144C, 7% VAF) in patient HES15. Rare CH detection was limited by the expected low frequency of expanded clones. With a median sequencing depth of 91, we achieved 84% power to detect variants with 5% VAF and 52% power to detect variants with 3% VAF.
Next, we performed a custom sex-chromosome analysis to detect somatic variants that could explain the male bias of PDGFR-associated HESN, as have been associated with other sex-biased cancers.21-23 No broad or focal copy number changes targeting specific genes were found on chromosome X (chrX) or chrY (Figure 2E, supplemental Figure 4). Importantly, no clonal loss of chrY was observed, as previously described in leukocytes of aging men.24 There were no recurrent point mutations in protein-coding genes or regulatory elements, including in tumor suppressors that escape chrX inactivation21 or their homologs on chrY.
Here, we show that PDGFR-rearranged HESN has simple genomics. We did not observe any frequent or recurrent cooperating mutations, including in known cancer genes or in genes associated with age-related CH. Most of the tumor samples demonstrated a linear phylogeny, and the PDGFR rearrangement was truncal (part of the founding clone) in all cases. This may explain why patients have a robust and sustained response to kinase inhibitors.
There was no obvious genetic feature that explains the extreme male bias of F/P-associated HESN. It is possible that hormonal or immune environment differences predispose men to the disease, or conversely, protect women from the disease. Alternatively, dosage imbalances in the X or Y genes among men and women, even in the absence of a mutation, may predispose eosinophils to acquire PDGFR rearrangements or exert positive selection on mutant cells. Future work using male and female experimental systems that model eosinophil transformation may address these questions.
There are important limitations to this work. Firstly, in this rare disease, the sample size was limited despite recruiting from a major referral center for eosinophilic disorders. This constrained our ability to identify events occurring in a subset of patients, and severely limited the identification of germ line predisposition. Secondly, the WGS-read depth was limited and might have missed subclonal events. Thirdly, the control samples were remission peripheral blood cells, which could have caused us to miss detecting a mutation acquired in all hematopoietic cells that predisposes to HESN. To address this, we manually curated all somatic variant calls in the normal samples for genes known to be associated with CH or blood cancer. However, this might have missed novel predisposing mutations. These limitations can be overcome in a future study using nonhematopoietic normal tissue as the reference. Thus, we conclude that neoplastic HES has linear genomics, with the PDGFR rearrangement as the only unambiguous recurrent genetic driver event.
Acknowledgments: The authors thank Chip Stewart, Elizabeth Martin, Ignaty Leshchiner, Jason Nomburg, and Kirsten Kubler for analysis support and Alexis Berry and Sana Mahmood for assistance in preparing the DNA samples. This work was financially supported by the Mark Foundation For Cancer Research (A.A.L.) and the Bertarelli Rare Cancers Fund at Harvard Medical School (A.A.L. and E.R.). A.A.L. is a scholar of the Leukemia & Lymphoma Society. This work was also supported in part by the Division of Intramural Research, The National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Contribution: E.R., A.D.K., and A.A.L. contributed toward the study design, data analysis, manuscript preparation, and editing; M.Q., J.M.B., A.J.O., L.T., M.M., and I.M. performed experiments, data analysis, and manuscript editing.
Conflict-of-interest disclosure: A.A.L. is a consultant for Qiagen and has received research support from AbbVie and Stemline Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Esther Rheinbay, Broad Institute of Harvard and MIT and Massachusetts General Hospital Cancer Center, Harvard Medical School; e-mail: erheinbay@mgh.harvard.edu; and Andrew A. Lane, Broad Institute of Harvard and MIT and Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School; e-mail: andrew_lane@dfci.harvard.edu.

