

Abstract
Abstract 3011
Poster Board II-987
Somatic mutation of the X- linked gene PIG-A results in partial or absolute deficiency of cell surface expression of glycosylphosphatidylinositol-anchored proteins (GPI-APs). In humans, this genetic event in a hematopoietic stem cell leads to the blood disease paroxysmal nocturnal hemoglobinuria (PNH). PNH has two major clinical presentations: predominantly hemolytic without overt marrow failure, referred to as classic PNH, and PNH clonal expansion in the setting of marrow failure, or the aplastic anemia/PNH syndrome (AA/PNH). The diagnosis of PNH is based on the presence of GPI-AP-deficient clones mainly within erythrocytes and granulocytes. Since PNH is a stem cell disorder, all hematopoietic lineages are involved and GPI-AP-deficient clones are found among platelets, monocytes and lymphocytes. Except for their different clinical presentations, classic PNH and AA-PNH have never been shown to have specific discriminating biological features. Indeed, we previously reported no difference in the expression profile of CD34 cells in hemolytic and aplastic forms of PNH (Chen G et al, Leukemia 2005; 19:862). As marrow failure in PNH, as in aplastic anemia, is immune-mediated, and T cells are affected by the PIG-A mutation, we hypothesized that the mutant subset of lymphocytes might differ in PNH. We compared by microarray the T cell compartment in both forms of the disease. We sorted peripheral blood GPI-AP-normal and -deficient pooled CD4 and CD8 T cells using CD59 and CD55 as GPI-AP markers, and subjected each subset to array hybridization and analysis. Samples were obtained from 1 patient with classic PNH, 3 patients with AA-PNH syndrome and 3 age-matched normal donors as controls. In preliminary control experiments, we determined using microarray of total RNA derived from healthy donors' T cells, with and without GPI-AP-specific antibodies, the absence of altered gene expression pattern or activation of specific genes due to selective antibody binding to normal cells. By principal component analysis (PCA), the phenotype of the PNH clone in classic PNH was divergent from the AA-PNH clone and from that of normal donors. By consistency test, only 15 probesets/genes resulted common in the comparison between GPI-AP-deficient and GPI-AP-normal cells between classic PNH and AA-PNH syndrome. Using Ingenuity Software, we found that the represented probesets/genes were involved in some major canonical pathways such as cell death, immune and lymphatic system development, immune response, and cell-to-cell signaling interaction. In order to investigate the different gene expression profile derived from specific subset of T cells, we sorted GPI-AP-deficient and GPI-AP-normal CD8 T cells using CD48 and CD52 as markers. When we compared GPI-AP-deficient and GPI-AP-normal cells between classic PNH and AA-PNH syndrome, we found that only 23 probesets/genes were up-regulated and 132 down-regulated confirming the divergent gene expression profile between the two diseases (Figure 1). We conclude that the restricted similarity in the gene expression profile in the GPI-AP-deficient T cells between classic PNH and AA-PNH syndrome leads to a specific “signature” of genes that might support the different clinical presentation and evolution of the two diseases.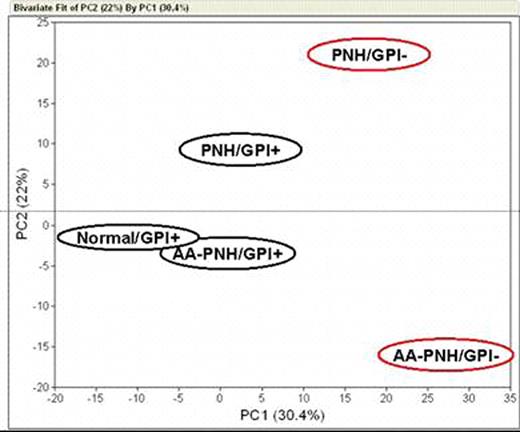 Close modal
Close modal
Figure 1.

Divergent gene expression profile between GPI-AP-deficient and GPI-AP-normal cells in classic PNH and AA-PNH syndrome
Figure 1.
Divergent gene expression profile between GPI-AP-deficient and GPI-AP-normal cells in classic PNH and AA-PNH syndrome
Disclosures:
No relevant conflicts of interest to declare.

