

Key Points
FCA is a novel flow cytometry–based platelet aggregation assay that allows single receptor analysis in small volume/thrombocytopenic samples
FCA facilitates platelet studies in experimental animal models even during gestation and allows kinetic measurements in individual animals
Abstract
The main function of platelets is to maintain normal hemostasis. Inefficient platelet production and/or defective platelet function results in bleeding disorders resulting from a wide range of genetic traits and acquired pathologies. Several platelet function tests have been developed for use in the clinic and in experimental animal models. In particular, platelet aggregation is routinely measured in an aggregometer, which requires normal platelet counts and significant blood sample volumes. For this reason, the analysis of thrombocytopenic patients, infants, and animal models is problematic. We have developed a novel flow cytometry test of platelet aggregation, in which 10- to 25-fold lower platelet counts or sample volumes can be used, either of platelet-rich plasma or whole blood from human subjects or mice. This setup can be applied to test in small assay volumes the influence of a variety of stimuli, drugs, and plasma factors, such as antibodies, on platelet aggregation. The presented principle stands as a novel promising tool, which allows analysis of platelet aggregation in thrombocytopenic patients or infants, and facilitates studies in platelets obtained from experimental animal models without the need of special devices but a flow cytometer.
Introduction
Platelets are the blood cells responsible for maintaining hemostasis. In addition, more recently, platelets have also been implicated, among others, in the regulation of immune responses, cancer metastasis, vascular development, and angiogenesis.1-6 Different aspects of platelet function relevant for hemostasis (ie, platelet adhesion, activation, and aggregation) can be tested in vitro.7,8 Adhesion and activation parameters can be extrapolated from the analysis of relevant plasma proteins, surface marker expression, granular content and release, and signaling pathways.7,8 However, the only truly functional platelet tests used to date, such as the bleeding time (which is losing popularity)9-11 and light transmission aggregometry (LTA), have major methodological limitations.8,12-19
LTA relies on the measurement of light transmission through a cuvette filled with platelet-rich plasma (PRP) placed between a light source and a photocell. This test is based on the decrease in light scattering upon addition of a platelet agonist resulting from the increased number of platelet aggregates. As a result, the turbidity of the sample decreases and the light transmission detected by the photocell increases.12,13 In view of the variations in methodology and output, efforts have been undertaken to standardize LTA conditions.14,15 Based on these comparative studies, it was recommended to perform the test with freshly isolated PRP and not to adjust the platelet counts. However, a negative result of a standard LTA on thrombocytopenic patients with PRP platelet numbers below 50 × 106/mL deviates from normal and cannot be interpreted as indicative for a defect in platelet aggregation. LTA therefore requires a significant sample volume to obtain enough PRP to be tested.16-18 This renders the analysis of samples from infants suspected of platelet aggregation defects very difficult. It is also not suitable for use in lipemic blood samples.8,19 Moreover, to perform 1 aggregometry test with mouse blood with the conventional aggregometer, a single mouse has to be euthanized to obtain enough material.20 Kinetic studies in the same animal are thus impossible. Furthermore, LTA requires a specific device to measure aggregation. The generalized use of citrate as anticoagulant has an additional drawback, because due to the calcium ion concentration changes, the response to certain agonists increases.8 In general, LTA remains poorly reproducible and standardized between different laboratories,14-16,21 is labor intensive, and does not simulate primary hemostasis because it uses PRP instead of whole blood.8
Platelet aggregation analysis within diluted whole blood is possible with impedance aggregometry. This method is based on the binding of platelets to 2 electrodes immersed in the stirring sample when a current is passed through it. When an agonist is added, more platelets will bind to the electrodes, and the extent and increase in impedance is measured and correlated with platelet aggregation. Although this method can be used in lipemic blood samples, it is not suitable for use with thrombocytopenic patient material.19 Clearly, there is a demand for a functional test that does not suffer from these limitations.
We have developed a novel flow cytometry assay that allows the detection of platelet aggregates with small blood volumes and low platelet numbers. In brief, 2 portions of platelets from the same donor either washed or within the whole blood context are labeled with different fluorochromes and, after mixing and upon appropriate stimulation, analyzed for the formation of double-colored aggregates by flow cytometry. The protocol allows the detection of small aggregates after agonist incubation, as opposed to the gross aggregate formation required for LTA. This characteristic allows sensitive dissection of platelet receptor function when combining agonists and antagonists in the assay. In addition, the assay renders studies on the influence of plasma and/or drugs on platelet aggregation more straightforward and affordable. We also present a whole blood aggregation option that can be used with very small blood volumes. In combination with conventional surface marker analysis by flow cytometry and classical assays, this method represents a highly promising tool for platelet function tests in human as well as in animal models without the requirement for specialized equipment other than a flow cytometer.
Materials and methods
Preparation of human washed platelets
Whole blood collected in EDTA, heparin, or citrate was centrifuged for 15 minutes at 210 g, and the PRP fraction was collected. Platelets were washed twice with sequestrine buffer (17.5 mM Na2HPO4, 8.9 mM Na2EDTA, 154 mM NaCl, pH 6.9, containing 0.1% [wt/vol] bovine serum albumin) by centrifuging them 5 minutes at 2310g. Platelets were resuspended to a final concentration of 50 × 106 plt/mL in HEPES medium (132 mM NaCl, 6 mM KCl, 1 mM MgSO4, 1.2 mM KH2PO4, 20 mM HEPES, pH 7.4, containing 5 mM glucose). The study was approved by our institute medical ethics committee in accordance with the 1964 Declaration of Helsinki.
Labeling human washed platelets
Platelets were labeled with 0.1 µM carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR) or with 1.6 µM PKH26 (Sigma-Aldrich, St. Louis, MO) for 15 minutes at room temperature. The labeling was stopped by adding pooled CPD plasma from healthy AB+ donors to a final concentration of 20% (vol/vol).
Labeling human platelets in whole blood
Whole blood was incubated with 1:100 dilution of CD31-Pacific Blue (eBiosciences, San Diego, CA) or CD31-FITC (BD Biosciences, San Jose, NJ) for 15 min at room temperature. After washing twice with 2× volume sequestrine buffer + 0.1% bovine serum albumin 5 minutes at 2250g, whole blood was resuspended to the same starting volume in HEPES medium containing 20% (vol/vol) AB+ pool CPD plasma.
Preparation of mouse washed platelets and collection of embryonic blood
Mouse whole blood was taken by orbital, cheek, or heart puncture and collected in heparinized mouse blood collection tubes. The whole blood was diluted with 2× volume HEPES medium. Diluted blood was centrifuged 15 minutes at 50g, and the collected PRP was diluted further in HEPES medium to a final concentration of 50 × 106 plt/mL.
Mouse embryonic blood was collected as described: the blood of 8 to 15 13.5 days post coitum embryos was collected in approximately 8 mL of heparin–phosphate-buffered saline and filtered through a 40-µm cell strainer.22 All animal experiments were approved by the local ethical review committee.
Staining of mouse platelets in PRP and whole blood
Mouse PRP or whole blood was incubated with 1:100 dilution of CD9-APC and CD31-PECy7 or CD9-PE monoclonal antibodies (moAbs) (Abcam, Cambridge, UK) for 15 minutes at room temperature. In whole blood samples, an additional staining with 1:100 Ter119-PerCP-Cy5.5 (Ly-76) (eBiosciences) was performed to stain definitive erythrocytes (optional). When staining embryonic blood, antibodies were used in a 1:1000 concentration in a volume of 4 mL. After incubation, samples were centrifuged 5 minutes at 2250g (2× for whole blood samples) and resuspended in HEPES medium supplemented with 2% (vol/vol) heparin mouse plasma or 2% (vol/vol) AB+ pool CPD plasma to a concentration of 50 × 106 plt/mL in the case of PRP, and with 5% to 10% plasma to 100 to 300 × 106 plt/mL in the case of whole blood or embryonic blood.
Flow cytometry–based platelet aggregation assay
Preincubation of human samples.
Differently labeled washed platelets or whole blood samples were mixed 1:1, and 0.1× volume of healthy donor AB+ pool CPD plasma, supplemented with 20 µM D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (thrombin inhibitor, PPACK; Calbiochem, Merck Group, Darmstadt, Germany) was added to citrate anticoagulated samples. Alternatively, EDTA anticoagulated samples were supplemented with 1× volume healthy or patient EDTA plasma, 1 mM MgCl2, and 20 µM PPACK. Samples were incubated with or without antagonists at 37°C while shaking at 700 rpm for 15 minutes. As antagonists, we used 0.5 µg/mL tirofiban (αIIbβ3 integrin inhibitor Aggrastat; Merck, Whitehouse Station, NJ) or 10 µg/mL moAb LIA 1/2.1 (β1 integrin chain blocking moAb).23 After the incubation, 3 mM CaCl2 was added when performing the assay with washed platelets.
Preincubation of mouse samples.
The 2 populations of labeled washed platelets or whole blood were mixed 1:1 and then preincubated for 15 min at 37°C while shaking at 600 rpm. As antagonists we used 5 µg/mL tirofiban (Aggrastat), 20 µg/mL CD29 β1 integrin blocking moAb (Ha2/5; BD Pharmingen, San Diego, CA), 12.5µM Bay (Syk inhibitor IV; Bay-61-3606; Calbiochem), and 5 µg/mL JAQ1 (GPVI blocking moAb, M011-0; Emfret Analytics, Eibelstadt, Germany).
Activation.
Preincubated human platelets were activated with 100 ng/mL phorbol myristate acetate (PMA, Sigma-Aldrich), 10 µg/mL type-I collagen (Horm; Nycomed Arzneimittel GmbH, München, Germany), 300 µM Aggretin A,24 or 1.5 mg/mL ristocetin (Biopool; Trinity Biotech Plc, Bray, Co Wincklow, Ireland) at 37°C while shaking at 1000 rpm. Preincubated mouse platelets were activated with 10 µg/mL type-I collagen or 10 µg/mL botrocetin (Sigma-Aldrich) at 37°C while shaking at 1000 rpm or with 100 ng/mL PMA or 75 to 300 µM Aggretin A at 37°C while shaking at 700 rpm. At different times, samples were fixed by addition of 9× volume of 0.5% (vol/vol) formaldehyde (Polysciences Inc., Warrington, NJ, methanol-free) in phosphate-buffered saline.
Measurement.
Fixed samples were measured by flow cytometry (LSRII + HTS, BD Biosciences) and analyzed with BD FACS DIVA software (BD Biosciences) or FlowJo version 9.2 software (Tree Star, inc). Alternatively, samples were measured with the ImageStreamX (Amnis, Seattle, WA).
Quantification.
For analysis, a quadrant was set in the dot plot of respective channels on non-stimulated platelets. The appearance of double-colored events in the upper right quadrant (Q2) was quantified as percentage of total amount of labeled events (Q1+Q2+Q4) at every time point analyzed:
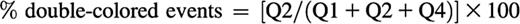
The decrease in total single-colored events at a given time point relative to the t = 0 sample can be calculated as follows and is an indirect measurement of the aggregation of platelets:
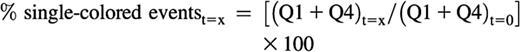
The average size of the aggregates at a given time point can be calculated as follows, although this is a relative or indicative measurement, and should not be taken into consideration when the Q2 event number falls below 100 or when the sum of single-colored events at a given time is higher than the sum of the single events at t = 0:
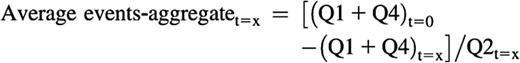
Measurements and calculations were made with mouse samples as described previously after gating out Ter119+ cells or including all cells in the analysis.
Light transmission aggregometry
Platelets were centrifuged for 5 minutes at 2310g and resuspended to a final concentration of 25 × 107 plt/mL in 40% (vol/vol) HEPES medium, 10% (vol/vol) CPD, and 50% (vol/vol) EDTA healthy donor plasma, supplemented with 1 mM MgCl2 and 20 µM PPACK. The stained and unstained platelets were incubated 15 minutes at 37°C while shaking at 600 rpm. A Lumi-aggregation module Series 1000B (Payton Scientific, Buffalo, NY) was attached to a linear recorder and set on slight stirring (800 rpm) at 37°C. The baseline was set with 450 µL of 40% (vol/vol) HEPES medium, 10% (vol/vol) CPD, and 50% (vol/vol) EDTA plasma. Cuvettes containing 450 µL of stained or unstained samples were measured and after stabilization of the signal, 3 mM CaCl2 was added. After restabilization of the signal, incubations were activated with either 100 ng/mL PMA or 10 µg/mL type-I collagen and light transmission was recorded over time.
Statistical analysis
Two-tailed Welch’s t test analysis (2 samples unequal variance) was performed when indicated.
Results
Labeling of platelets does not affect platelet morphology or function
To enable measurement of platelet aggregation by detection of double-colored events in a flow cytometer, human or mouse platelets were labeled with two different fluorochromes, either CFSE (a green fluorescent dye labeling the cytosol) and PKH26 (a red fluorescent dye labeling the plasma membrane) or with conjugated antibodies. The conditions of these labeling protocols did not affect the scatter characteristics or the morphology of the platelets as judged by light microscopy of formaldehyde-fixed samples (data not shown).
To exclude a possible effect of the labeling procedure on platelet aggregation, labeled platelets were employed in a standard LTA assay and their response toward collagen was compared with that of unlabeled platelets. The labeling with the 2 fluorescent dyes or with conjugated antibodies had a negligible effect on human and mouse platelet aggregation (Figure 1A). Therefore we proceeded with this labeling option to develop our flow cytometry-based platelet aggregation assay (FCA).

Effect of fluorescent labeling on platelet aggregation. (A) Light transmission aggregometry upon collagen stimulation of human platelets unlabeled or labeled with PKH26/CFSE (left), CD31 (middle), or mouse platelets unlabeled or labeled with CD9 (right). (B) Simplified scheme of FCA.
Effect of fluorescent labeling on platelet aggregation. (A) Light transmission aggregometry upon collagen stimulation of human platelets unlabeled or labeled with PKH26/CFSE (left), CD31 (middle), or mouse platelets unlabeled or labeled with CD9 (right). (B) Simplified scheme of FCA.
FCA allows assessment of single receptor function in samples with low starting volume or platelet number
We first isolated platelets from PRP, labeled them with either CFSE or PKH26, and mixed them 1:1 as described in “Materials and methods.” Platelet mixtures were stimulated and fixed at different times to measure the kinetics of aggregate formation by flow cytometry (Figure 1B). Upon stimulation with PMA (which activates the fibrinogen receptor αIIbβ3 integrin), the percentage of double-colored events (Figure 2A, Q2) increased over time and their forward scatter/side scatter positioning shifted compared with single platelets (Figure 2A). We measured with ImageStreamX our aggregation time series to visualize the single-colored platelets or double-colored aggregates (Figure 2B), which are small (2 to 15 platelets on average, data not shown, and supplemental Table 1). This characteristic made us investigate whether our test would allow studying the contribution of single receptors at the onset of aggregate formation.

Human platelet aggregation test with washed platelets. (A) Dot plots depicting platelets labeled with CFSE or PKH26 mixed in 1:1 ratio as measured by flow cytometry before addition of PMA (t = 0 min, left) or upon PMA stimulation (t = 10 min, right). Platelets stained with PKH26 are enclosed in quadrant Q1. Platelets stained with CFSE are enclosed in quadrant Q4. Double-colored events in quadrant Q2 represent aggregates before and after stimulation by PMA. All populations are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelet. (B) Images of single-stained platelets from quadrants Q1 and Q4 or aggregates from quadrant Q2 obtained with ImageStreamX are depicted next to the respective dot plots. BF, bright field. (C) Platelets were labeled with CFSE or PKH26 and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, or ristocetin alone or in the presence of either the integrin β3 inhibitor tirofiban or blocking anti-integrin β1 antibody LIA 1/2.1. Average and standard error of the mean are depicted, n > 3. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05).
Human platelet aggregation test with washed platelets. (A) Dot plots depicting platelets labeled with CFSE or PKH26 mixed in 1:1 ratio as measured by flow cytometry before addition of PMA (t = 0 min, left) or upon PMA stimulation (t = 10 min, right). Platelets stained with PKH26 are enclosed in quadrant Q1. Platelets stained with CFSE are enclosed in quadrant Q4. Double-colored events in quadrant Q2 represent aggregates before and after stimulation by PMA. All populations are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelet. (B) Images of single-stained platelets from quadrants Q1 and Q4 or aggregates from quadrant Q2 obtained with ImageStreamX are depicted next to the respective dot plots. BF, bright field. (C) Platelets were labeled with CFSE or PKH26 and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, or ristocetin alone or in the presence of either the integrin β3 inhibitor tirofiban or blocking anti-integrin β1 antibody LIA 1/2.1. Average and standard error of the mean are depicted, n > 3. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05).
We used a combination of stimuli and antagonists well-characterized for their specificity. As stimuli we used PMA, collagen (which binds to both α2β1 integrin and GPVI, which will in turn activate αIIbβ3 and α2β1 integrins),25,26 Aggretin A (which binds to CLEC-2 and will in turn activate αIIbβ3 integrin through a Syk-dependent mechanism),24,27,28 and ristocetin (which binds to and changes the conformation of von Willebrand factor-A, subsequently activating glycoprotein [GP] Ib). When using antagonists, we preincubated the platelets with tirofiban, a peptide antagonist of αIIbβ3 integrin, or LIA, a moAb directed against β1 integrin.23,29 As expected, tirofiban completely inhibited aggregate formation induced by PMA and Aggretin A, whereas LIA did not interfere with aggregation (Figure 2C) because PMA and Aggretin A both induce αIIbβ3-dependent aggregation. The decrease in single-colored events and the average size of aggregates remained unaltered in the presence of LIA (supplemental Table 1). In case of collagen stimulation, tirofiban did not exert an inhibitory effect on the aggregation kinetics, whereas LIA decreased by 75% the collagen-mediated aggregation (Figure 2C). However, tirofiban preincubation affected the size of aggregates and consequently the reduction in single-colored events (supplemental Table 1). This suggests that collagen-mediated aggregation requires functional α2β1 instead of αIIbβ3 integrin at the onset of aggregation and that αIIbβ3 integrin contributes to aggregate size and stability in a second wave. Aggregation induced by ristocetin was not inhibited by either tirofiban or LIA; neither were the aggregate size nor the decrease in single-colored events affected in the presence of the antagonists used (Figure 2C and supplemental Table 1). In conclusion, FCA of human washed platelets allows, in comparison with LTA, a distinction between α2β1 and αIIbβ3 integrin function that can be dissected by stimulating the platelets with either collagen (α2β1-mediated) or PMA (αIIbβ3-mediated), a feature that we have shown previously to distinguish the aggregopathy of Glanzmann thrombasthenia and leukocyte adhesion deficiency (LAD)-III patient-derived platelets.30,31
FCA allows testing platelet function in small volumes of whole blood
We next set out to optimize FCA for whole blood samples, with the intention to analyze platelet function in small volumes of whole blood. This is necessary when the clinical profile does not allow sampling the large volumes of blood required for proper PRP collection, as occurs with newborns, infants, or thrombocytopenic patients. For this purpose, we used antibodies directed against surface markers present on platelets, but not on red cells, that will not interfere with platelet aggregation. After testing several possibilities (data not shown), we selected CD31 as specified in “Materials and methods.” In Figure 3, we show representative data of whole blood platelet aggregation kinetics when using CD31-FITC and CD31-Pacific Blue as antibodies.

Human platelet aggregation test with whole blood. Platelets were labeled with CD31-Pacific Blue or CD31-FITC and mixed in 1:1 proportion prior to stimulation. (A) Representative dot plots before and after platelet aggregation upon stimulation with ristocetin. All populations (unstained, single-stained platelets and double-colored aggregates) are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelet; RBC, red blood cell. (B) Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, and ristocetin. Average and standard error of the mean are depicted, n > 3. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05). (C) Whole blood platelet aggregation analysis performed with blood from Glanzmann thrombasthenia and LAD-III patients upon collagen, PMA, or ristocetin stimulation. Note that Glanzmann platelets respond about half normally to collagen but not to PMA, whereas in LAD-III patients, collagen and PMA-induced aggregation is equally reduced. Control platelets were incubated with tirofiban (β3 integrin inhibitor) or tirofiban + LIA 1/2.1 (integrin β1 blocking moAb) to mimic the phenotype of Glanzmann thrombasthenia and LAD-III variant patients, respectively.
Human platelet aggregation test with whole blood. Platelets were labeled with CD31-Pacific Blue or CD31-FITC and mixed in 1:1 proportion prior to stimulation. (A) Representative dot plots before and after platelet aggregation upon stimulation with ristocetin. All populations (unstained, single-stained platelets and double-colored aggregates) are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelet; RBC, red blood cell. (B) Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, and ristocetin. Average and standard error of the mean are depicted, n > 3. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05). (C) Whole blood platelet aggregation analysis performed with blood from Glanzmann thrombasthenia and LAD-III patients upon collagen, PMA, or ristocetin stimulation. Note that Glanzmann platelets respond about half normally to collagen but not to PMA, whereas in LAD-III patients, collagen and PMA-induced aggregation is equally reduced. Control platelets were incubated with tirofiban (β3 integrin inhibitor) or tirofiban + LIA 1/2.1 (integrin β1 blocking moAb) to mimic the phenotype of Glanzmann thrombasthenia and LAD-III variant patients, respectively.
Upon stimulation with ristocetin, the percentage of double-colored events (Figure 3A, Q2) increased over time, and their forward scatter/side scatter positioning shifted compared with single platelets (Figure 3A). Red blood cells were detected in Q3, where other cells and plasma components are also distributed (Figure 3A).
Stimulation with PMA, collagen, Aggretin A, and ristocetin resulted in detectable platelet aggregation measured either directly (increase in double-colored events relative to total colored events, Figure 3B) or indirectly (decrease in single-colored events, supplemental Table 2). The aggregate size (events/aggregate) is equal (ristocetin) or larger with the same stimulus when performing the aggregation assay with whole blood in comparison with washed platelets (supplemental Tables 1 and 2).
We have previously shown that FCA allows the distinction between the aggregopathy of Glanzmann thrombasthenia vs LAD-III patients in washed platelets,31 based on the collagen-induced α2β1-dependent aggregation present in Glanzmann but absent in LAD-III platelets. Therefore, we aimed to validate our method in whole blood samples from Glanzmann thrombasthenia and LAD-III patients. As shown in Figure 3C, platelets from Glanzmann thrombasthenia and LAD-III patients respond normally to stimulation by ristocetin as compared with platelets from healthy controls, set to 100%. LAD-III platelets have an equally reduced response to collagen and PMA, whereas Glanzmann platelets have a better response to collagen than to PMA (Figure 3C and supplemental Table 2), confirming previous results and suggesting that the still-functional α2β1 integrin in Glanzmann platelets could explain the less severe bleeding phenotype of Glanzmann patients when compared with LAD-III patients.30,31
FCA allows testing the effect of platelet autoantibodies in plasma
There is a plethora of conditions in which platelet autoantibodies are generated or induced in a patient. It is relevant to the clinic to know not only the molecule recognized by the autoantibodies, but also to test their effect on platelet function. We have tested the plasma of immune thrombocytopenic purpura patients with persisting skin bleedings after splenectomy in spite of increased platelet counts and compared them with platelets from healthy donors. These patients produce strong reactive autoantibodies against αIIbβ3 integrin, which might result in altered platelet function.
We used ristocetin as a αIIbβ3-independent agonist as a positive control for the quality of the plasma and the platelets. As can be seen, most of the plasma samples induced a comparable or even a better response than did the healthy plasma controls, which were set to 100%. The increased response to ristocetin in some of these patient plasma samples (Figure 4, left) could be explained by increased levels of von Willebrand factor (data not shown), as occurs generally in thrombocytopenic patients.32-34 When stimulated with PMA, 2 patient plasma samples reduced aggregation by 10%, 2 patient plasma samples reduced aggregation by 35%, and 1 patient’s plasma did not affect the PMA-induced aggregation (Figure 4, right). One of the plasmas slightly increased and another decreased up to 80% both responses. The inhibitory effect of αIIbβ3 integrin autoantibodies on platelet aggregation is known for acquired Glanzmann thrombasthenia patients, with a primary coagulation defect in spite of (near) normal platelet counts.35 This inhibitory effect exists in some immune thrombocytopenic purpura patients that, next to the thrombocytopenia, further impairs the primary coagulation. Clearly, this effect can be characterized by FCA.

Effect of plasma on human platelet aggregation test. The plasma from patients with confirmed αIIbβ3 integrin autoantibodies was used with matched blood group, healthy donor platelets labeled with either CFSE or PKH26 and mixed in 1:1 ratio before stimulation. Ristocetin stimulation (left) was used as a control of the plasma quality because GPIb-induced platelet aggregation is independent of αIIbβ3 integrin function. PMA stimulation (right) was used to test the potential inhibitory effects of patient autoantibodies on αIIbβ3 integrin function. Aggregation percentages were normalized to those obtained with the same platelets incubated with healthy EDTA pool plasma (set to 100%) and either agonist. Average and standard deviation are depicted for the group of control plasmas on each condition, n = 4. Some of the patient plasmas were tested in 2 independent experiments.
Effect of plasma on human platelet aggregation test. The plasma from patients with confirmed αIIbβ3 integrin autoantibodies was used with matched blood group, healthy donor platelets labeled with either CFSE or PKH26 and mixed in 1:1 ratio before stimulation. Ristocetin stimulation (left) was used as a control of the plasma quality because GPIb-induced platelet aggregation is independent of αIIbβ3 integrin function. PMA stimulation (right) was used to test the potential inhibitory effects of patient autoantibodies on αIIbβ3 integrin function. Aggregation percentages were normalized to those obtained with the same platelets incubated with healthy EDTA pool plasma (set to 100%) and either agonist. Average and standard deviation are depicted for the group of control plasmas on each condition, n = 4. Some of the patient plasmas were tested in 2 independent experiments.
FCA allows testing small volumes of mouse PRP and adult and embryonic whole blood
Mouse models are increasingly used to unravel platelet function mechanisms at the genetic level or in preclinical studies. However, assessing platelet function in small rodents requires in most cases euthanizing an animal for a single test. This precludes kinetic measurements in individual animals. For the same reason, the analysis of platelet function during the mouse fetal stages is very difficult. We therefore set out to validate our FCA method with mouse washed platelets, whole blood, and embryonic blood.
We tested mouse platelet aggregation upon PMA, collagen, Aggretin A, and botrocetin (which induces platelet aggregation in mice by activating GPIb, as does ristocetin with human platelets), and we employed several antagonists to each stimulation, ie, tirofiban, CD29 (a β1 integrin blocking moAb), Bay (Syk inhibitor), and JAQ1 (GPVI inhibitor) (Figure 5 and supplemental Table 3). As shown in Figure 5A, PMA-induced aggregation was only inhibited by tirofiban, which confirms that this stimulus is αIIbβ3-dependent. Collagen-induced aggregation depends on functional GPVI and α2β1 integrin, Syk signaling, and, as opposed to human platelets in this assay, it depends on functional αIIbβ3 integrin (Figure 5B). Aggretin A–induced aggregation was dependent on functional αIIbβ3 integrin and Syk signaling, as has been previously described (Figure 5C,E).24,28 Botrocetin-induced aggregation was in our assay not influenced by any of the antagonists used (Figure 5D). Figure 5E depicts an example of the aggregation kinetics upon stimulation with Aggretin A: the percentage of double-colored events (Q2) increased over time and their forward scatter/side scatter positioning shifted compared with single platelets.

Mouse platelet aggregation test with washed platelets. Platelets were labeled with CD9-PE or CD9-APC and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA (A), collagen (B), Aggretin A (C), and botrocetin (D). Average and standard error of the mean are depicted when indicated, n > 3. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05). Representative data out of at least 5 independent experiments are depicted. We combined the agonists with the following antagonists: tirofiban (integrin β3 inhibitor), blocking anti-CD29 antibody (integrin β1), Bay (Syk inhibitor), and JAQ1 (GPVI blocking antibody). (E) An example of the gating used after flow cytometry analysis is depicted. Note the increase in events in Q2 after stimulation with Aggretin A. All populations are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelets.
Mouse platelet aggregation test with washed platelets. Platelets were labeled with CD9-PE or CD9-APC and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA (A), collagen (B), Aggretin A (C), and botrocetin (D). Average and standard error of the mean are depicted when indicated, n > 3. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05). Representative data out of at least 5 independent experiments are depicted. We combined the agonists with the following antagonists: tirofiban (integrin β3 inhibitor), blocking anti-CD29 antibody (integrin β1), Bay (Syk inhibitor), and JAQ1 (GPVI blocking antibody). (E) An example of the gating used after flow cytometry analysis is depicted. Note the increase in events in Q2 after stimulation with Aggretin A. All populations are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelets.
Next, we aimed to detect mouse platelet aggregation in whole blood samples. Optionally, it is possible to exclude erythroid blood cells by using Ter119-PerCPCy5.5 antibodies and gating the Ter119- population before the analysis of double-colored events. Upon stimulation with collagen, the percentage of double-colored events (Figure 6A, Q2) increased over time and their forward scatter/side scatter positioning shifted compared with single platelets (Figure 6A). Red blood cells were detected in Q3, where other cells and plasma components are also included (Figure 6A). As shown in Figure 6B, mouse platelet aggregation induced by PMA, collagen, Aggretin A, and botrocetin can be reliably measured in whole blood. As occurs with human platelets, the average events/aggregate is larger with the same stimulus when the aggregation test is performed in whole blood than with PRP-derived platelets (supplemental Tables 3 and 4). Summarizing, FCA allows platelet function analysis with a very small volume of blood (ie, 40-80 µL of blood are needed for testing 4 agonists with 4 aggregation time points per agonist), thus allowing time studies within the same animal in the context of clinical treatments or developmental studies.

Mouse platelet aggregation test with whole blood. Platelets were labeled with CD9-APC or CD9-PE and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, and botrocetin. Average and standard error of the mean are depicted, n > 3. (A) An example of the gating strategy used after flow cytometry analysis. Ter119− cells were selected to calculate double-colored events by plotting them against PE/APC channels. The same dot plot is shown for total events. Note the increase in events in Q2 after stimulation with collagen. Q1-Q4 and RBC populations are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelets; RBC, red blood cells. (B) Time course of double-colored events upon stimulation with PMA, collagen, Aggretin A, and botrocetin. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05).
Mouse platelet aggregation test with whole blood. Platelets were labeled with CD9-APC or CD9-PE and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, and botrocetin. Average and standard error of the mean are depicted, n > 3. (A) An example of the gating strategy used after flow cytometry analysis. Ter119− cells were selected to calculate double-colored events by plotting them against PE/APC channels. The same dot plot is shown for total events. Note the increase in events in Q2 after stimulation with collagen. Q1-Q4 and RBC populations are plotted against the forward and side scatter and indicated by colored arrowheads. PLT, platelets; RBC, red blood cells. (B) Time course of double-colored events upon stimulation with PMA, collagen, Aggretin A, and botrocetin. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05).
Finally, we aimed to measure platelet aggregation of mouse embryonic platelets within the whole embryonic blood context. We collected 13.5 days post coitum embryonic blood, stained the platelets, and performed the aggregation test as described in “Materials and methods.” As shown in Figure 7, we could detect platelet aggregation when activating the platelets with the 4 agonists used (ie, PMA, collagen, Aggretin A, and botrocetin), although the response to collagen stimulation was very low. This low collagen response of embryonic platelets has been previously described in porcine and human platelets,36,37 and it might be occurring also with mouse platelets during ontogeny. Because the marker we use to label platelets (CD9) is also expressed by primitive erythroid cells (primitive red blood cells, Figure 7A), we performed an extra gating of CD9-positive cells (PE or APC gates) and selected single platelets as forward scatter low. We did not perform an equal selection from the Q2 gate of aggregates, because aggregates shift to higher forward scatter values. We conclude that FCA allows the study of platelet function during the ontogeny of the mouse (Figure 7 and supplemental Table 5), a clearly advantageous functional application when it is of interest to measure platelet aggregation defects in case of mouse models that do not get to term.

Mouse platelet aggregation test with embryonic whole blood. Platelets were labeled with CD9-APC or CD9-PE and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, and botrocetin. Average and standard error of the mean are depicted, n > 3. (A) An example of the gating strategy used after flow cytometry analysis. CD9-PE or CD9-APC positive cells (gates APC or PE) were plotted against the forward scatter to select platelets (forward scatter low, named Q1 or Q4 to follow the nomenclature used in the formulas given in “Materials and methods”) or primitive RBC that are also CD9-positive (forward scatter high, pRBC). Note the increase in events in Q2 after stimulation. Within the double-negative gate Q3, Ter119 marked dRBC and the rest were components of blood and plasma that are negative for all markers used (plasma). All these populations are plotted against the forward and side scatter and indicated by colored arrowheads. d, definitive; p, primitive; PLT, platelets; RBC, red blood cells. (B) Time course of double-colored events upon stimulation with PMA, collagen, Aggretin A, and botrocetin. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05).
Mouse platelet aggregation test with embryonic whole blood. Platelets were labeled with CD9-APC or CD9-PE and mixed in 1:1 ratio before stimulation. Aggregation was measured by flow cytometry in time after stimulation with PMA, collagen, Aggretin A, and botrocetin. Average and standard error of the mean are depicted, n > 3. (A) An example of the gating strategy used after flow cytometry analysis. CD9-PE or CD9-APC positive cells (gates APC or PE) were plotted against the forward scatter to select platelets (forward scatter low, named Q1 or Q4 to follow the nomenclature used in the formulas given in “Materials and methods”) or primitive RBC that are also CD9-positive (forward scatter high, pRBC). Note the increase in events in Q2 after stimulation. Within the double-negative gate Q3, Ter119 marked dRBC and the rest were components of blood and plasma that are negative for all markers used (plasma). All these populations are plotted against the forward and side scatter and indicated by colored arrowheads. d, definitive; p, primitive; PLT, platelets; RBC, red blood cells. (B) Time course of double-colored events upon stimulation with PMA, collagen, Aggretin A, and botrocetin. Nonstimulated labeled platelets are shown as baseline for spontaneous aggregation. Colored asterisks at each data point indicate statistical significance with the unstimulated control (P < .05).
Discussion
We have developed a sensitive test for platelet aggregation that we have named FCA. The protocol is based on the fluorescent labeling of platelets with 2 different dyes or fluorochrome-conjugated antibodies, and subsequent measurement of double-colored events on a flow cytometer, as adapted from previous aggregation assays of neutrophils and lymphocytes.38 The labeling procedure did not affect platelet aggregation as measured by LTA and as shown in equivalent unstimulated time point samples measured by FCA.
It should be noted that the percentage of double-colored events underestimates the true number of aggregated platelets, because it counts aggregates of ≥2 platelets as a single event, and it excludes small aggregates containing—by chance—platelets with 1 label only. The contribution of the first phenomenon can be estimated from the number of platelets disappearing from the single-colored population. This indirect calculation (single-colored events disappearing) can be useful when using high platelet counts, but also in whole blood FCA because platelet aggregation with other blood cell types can result in much bigger aggregates that are not detected. Nevertheless, when performing FCA with washed platelets, we recommend maintaining a low platelet concentration (10-50 × 106/mL), because it allows the study of individual receptor function with a larger time response window. Whole blood FCA works optimally with higher platelet numbers (up to 300 × 106/mL), although it is sensitive enough with counts as low as 10 × 106/mL.
When assaying the function of washed human platelets, the formation of double-colored events was completely inhibited by the peptide antagonist tirofiban when using PMA as stimulus, as expected. With collagen as stimulus, the aggregation response appeared not to be dependent on αIIbβ3 integrin, because this aggregation failed to be inhibited by tirofiban. This was an unexpected result because collagen is known to trigger multiple synergistic pathways via multiple receptors, which would merge on αIIbβ3 integrin activation, required for aggregate formation.39 On the other hand, the binding of soluble collagen to activated platelets has been shown to be dependent on α2β1 integrin.40 In support of this notion, we observed that the response after collagen stimulation in our assay was strongly inhibited by LIA, which is directed against α2β1 integrin. This antibody did not interfere with the aggregation responses elicited by PMA or ristocetin. As more specifically developed in a separate study,31 this particular characteristic brings about a novel feature of FCA: whereas the readout of classical aggregometry is dependent on αIIbβ3 integrin function upon collagen stimulus (ie, large aggregates), we can now distinguish between aggregation mediated by αIIbβ3 or α2β1 integrin because we are able to measure the early onset of aggregate formation. This feature allowed us to distinguish the aggregopathy accompanying Glanzmann thrombasthenia (αIIbβ3 integrin/fibrinogen receptor absent, α2β1 integrin functional) and the aggregopathy accompanying LAD-III (both integrins inactive), because we could still detect collagen-induced aggregates in Glanzmann thrombasthenia but not in LAD-III platelets.31 Furthermore, tirofiban preincubation affected the size of collagen-induced aggregates and consequently the reduction in single-colored events. This suggests that collagen-mediated aggregation requires functional α2β1 integrin instead of αIIbβ3 integrin at the onset of aggregation and that αIIbβ3 integrin contributes to aggregate size and stability in a second wave. Translated to a physiological context, collagen-induced FCA probably represents the platelet adhesion to and aggregation on the damaged blood vessel wall, which also involves primarily interaction with α2β1 followed by αIIbβ3 mediated amplification. Of note, whereas PMA, collagen, Aggretin A, and ristocetin can be used in 1- to 2-day-old blood samples, other agonists such as ADP or thrombin receptor agonist peptide 6 (data not shown) perform properly only in fresh samples.
In case of whole blood aggregation, we detected a higher percentage of double-colored events when compared with aggregation of washed platelets. In addition, we detected platelet–red cell aggregates and platelet–mononuclear cell aggregates, which contribute also to clot formation in vivo (data not shown). Thus, many more different parameters have to be taken into account when measuring platelet aggregation in whole blood than is the case with washed platelets. This is the reason why dissection of single receptor function is more straightforward in the washed platelet setup than in whole blood, because aggregation to other cell types might mask the function of a specific receptor at the onset of aggregate formation. We additionally have observed an important contribution of αIIbβ3 integrin in the collagen-induced aggregation process in the whole blood context (Figure 3C). In concordance with this, when assaying the function of Glanzmann thrombasthenia and LAD-III patients in whole blood, we could still distinguish between the two aggregopathies; however, Glanzmann platelets aggregated 50% of control platelets upon collagen stimulation, as opposed to the normal aggregation levels we detected in washed platelet samples.31
We have shown that FCA allows not only the detection of intrinsic defects in platelet aggregation, but can also be used to detect blocking autoantibodies present in plasma and directed against platelet membrane antigens. With the same philosophy, FCA can be used to test chemical libraries or drugs, in small volumes of multiple samples on a large scale, thereby reducing the costs. This is potentially very relevant for screening studies for the development of novel hemostatic drugs, but also for patient-directed antiplatelet therapy monitoring.41,42 In addition, FCA can be applied to study platelet aggregation induced by bacteria or bacteria–platelet binding in washed platelets or whole blood. Relevant in the context of transfusion monitoring, platelet–blood cell interactions can be also potentially measured by FCA.
We further validated FCA for use with mouse platelets, either washed or in the whole blood context, and also fetal platelets in the embryonic blood context. We used botrocetin instead of ristocetin to induce GPIb-mediated platelet aggregation, because ristocetin is known to be unable to induce mouse platelet agregation.43,44 We noted that collagen-induced aggregation always required αIIbβ3 integrin function in mouse platelets, either washed or in the whole blood context, in contrast to what we found in human platelets. This could be due to species evolutionary divergence.45 The β3-independent collagen-induced platelet aggregation in human washed platelets is a process that occurs during a very small time window at physiological conditions, and the signaling cascade toward αIIbβ3 integrin seems to be much faster in the mouse platelets. When analyzing the function of fetal platelets, we observed a very low response upon collagen stimulation, as has been described for human and porcine platelets.36,37 Thus, this “fetal platelet dysfunction” appears conserved among species.
In conclusion, we provide a novel flow cytometry-based platelet aggregation assay that we have termed FCA, which can be performed with small starting volumes or low numbers of platelets. In combination with conventional surface marker analysis by flow cytometry and classical assays, FCA stands as a promising tool for the diagnosis of platelet defects, for screening of drug effects on platelet function, and for studies in animal models.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to Dr F. Sánchez-Madrid (Universidad Autónoma de Madrid, Spain) for providing moAb LIA1/2.1, and to Dr P.W. Kamphuisen and Dr M. Peters (Academic Medical Centre, Amsterdam, The Netherlands), as well as Dr Ö. Sanal and Dr M. Çetin (Hacepette University, Ankara, Turkey) for providing patient material. The authors thank Prof. D. Roos for critically reading the manuscript.
This work was supported by the Landsteiner Foundation for Blood Transfusion Research (grant 0619) (T.W.K. and E.v.d.V.), Sanquin Blood Supply, grant number PPOC-08-004 (I.M.D.C.), the Center for Translational Molecular Medicine program Innovative Coagulation Diagnostics (I.M.D.C., M.M.), The Netherlands Organization for Scientific Research (863.09.012) (L.G.), and the Deutsche Forschungsgemeinschaft (Eb177/9-1) (J.A.E.).
Authorship
Contribution: I.M.D.C. and M.M. designed and performed experiments and analyzed data; E.v.d.V. performed experiments; D.d.K. designed experiments and wrote the paper; L.P., M.d.H., J.A.E., K.S., S.R., and D.P. provided material and participated in discussions; T.W.K. designed experiments and wrote the paper; A.J.V. designed experiments and wrote the paper; T.K.v.d.B. designed experiments and wrote the paper; and L.G. designed and performed experiments, analyzed data and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.J.V. is the Department of Medical Biochemistry, Academic Medical Centre, University of Amsterdam, The Netherlands.
Correspondence: Laura Gutiérrez, Department of Blood Cell Research, Sanquin Research and Landsteiner Laboratory, Academic Medical Center, University of Amsterdam, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: l.gutierrez@sanquin.nl.

