TO THE EDITOR:
Ibrutinib is a small molecule Bruton tyrosine kinase (Btk) inhibitor approved by the Food and Drug Administration for clinical use in the treatment of chronic lymphocytic leukemia, Waldenström macroglobulinemia, and as a second-line treatment of lymphoma and chronic graft-versus-host disease.1 An association with pulmonary aspergillosis was observed shortly after Ibrutinib was licensed for use.2 A recent phase Ib study of Ibrutinib treatment of primary central nervous system lymphoma reported a 39% incidence of invasive aspergillosis, in patients concurrently treated with corticosteroids, in the absence of neutropenia.3 Studies of Aspergillus fumigatus infection in Btk−/− mice revealed focal pneumonia and large airway mucous plugs, mirroring findings in macrophage-depleted models of pulmonary aspergillosis.3
We recently described a key role for Btk in macrophage immune responses during experimental pulmonary aspergillosis.4 Btk was critical for endosomal signaling responses during murine macrophage phagocytosis of A fumigatus. Btk activation led to calcineurin-NFAT signaling, which was crucial for orchestrating neutrophil recruitment during pulmonary aspergillosis and was dependent on the endosomal DNA receptor TLR9. These observations suggest that defects in macrophage Btk signaling contribute to susceptibility to pulmonary aspergillosis. Here we show that Ibrutinib is a potent inhibitor of both NFAT and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) responses in human macrophages during infection with A fumigatus. We show that A fumigatus induces human macrophage Btk phosphorylation, and that Btk depletion impairs NFAT and NF-κB responses in human macrophages. Our findings suggest Btk involvement in a TLR9-dependent endosomally driven pathway in accordance with previous findings in our murine model. In addition, our results show that Ibrutinib is a strong inhibitor of macrophage responses to A fumigates, which may increase the susceptibility of patients on Ibrutinib to invasive aspergillosis.
Peripheral blood samples were collected from unscreened healthy donors. THP-1–derived macrophages, human monocyte–derived macrophages (hMDMs), and alveolar macrophages were isolated and differentiated as previously described.5 The study was approved by the Biomedical Research Unit (National Research Ethics Service reference 10/H0504/9[DAJ3]), Royal Brompton, and Harefield NHS Trust.
A fumigatus strain CEA10 (FGSC A1163) and Candida albicans ATCC 90028 were obtained from the Fungal Genetics Stock Center. ATCC 46645-eGFP was a kind gift from Frank Ebel (Germany). Strains were cultured as previously described.5
Macrophages were incubated with 1 μM Ibrutinib (Selleck Chemicals), 10 μm ODN2088 (TLR9-blocking nucleotide), 10 μM ODN20958 (control nucleotide, Miltenyi Biotec), 50 μg/mL zymosan, or vehicle. SMARTpool siGENOME BTK small interfering RNA (siRNA; Dharmacon) was used at a concentration of 75 nM. For siRNA knockdown, primary monocyte cells were differentiated for 7 days. On day 4, siRNA was transfected using VIromer Blue (Lipocalyx) according to the manufacturer’s instructions. Scramble siRNA was used as a control for all experiments.
Confocal microscopy was performed as previously described.5 Cells were permeabilized in phosphate-buffered saline containing 10% goat serum and 0.1% Saponin (Sigma, UK) for 2 hours and then incubated overnight at 4°C with a primary antibody (anti-NFATc1, clone 7A6, BD Biosciences; anti-NF-κB p65, clone F6, Santa-Cruz Biotech; anti-BTK, clone 7F12H4, Novus Biologicals) in blocking buffer. After washing with phosphate-buffered saline, cells were incubated with anti-rabbit Cy5 or anti-mouse Cy5 antibody (Life Technologies) for 45 minutes at room temperature and mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories).
Tumor necrosis factor-α (TNF-α) release was quantified in culture supernatants using DuoSet ELISA Development kit (R&D Systems) following the manufacturer’s instructions. Galactomannan was quantified in supernatants using Platelia Aspergillus Ag kit (Bio-Rad) following the manufacturer’s instructions. Western blotting for nuclear and cytoplasmic fractions was performed as previously described.5 For Btk phosphorylation studies, macrophages were incubated in 100 μM sodium pervanadate for 2 hours at 4°C prior to cell lysis. Membranes were probed with anti-NFATc1 (7A6; Santa-Cruz), anti-NFkB p65 (C22B4), anti-HDAC1 (10E2), anti-histone H3 (D1H2), anti-phospho-BTK (Tyr 223), and anti-BTK (D3H5) antibodies, all from Cell Signaling.
To determine whether A fumigatus activates Btk macrophages, THP-1 macrophages were infected with swollen conidia and phosphorylation of Btk at Tyr 223 determined by western blotting (Figure 1A). Infection induced phosphorylation of Btk, which was blocked by Ibrutinib. In addition, Ibrutinib inhibited A fumigatus–dependent nuclear translocation of NFAT and NF-ĸB (Figure 1B). Zymosan, but not C albicans, was able to induce BTK phosphorylation (supplemental Figure 1, available on the Blood Web site). The role of Btk in NFAT and NF-ĸB translocation was confirmed by Btk siRNA knockdown during A fumigatus infection of hMDMs, by confocal microscopy (Figure 1C-D; supplemental Figure 2). Accordingly, both Ibrutinib and Btk siRNA inhibited hMDM and alveolar macrophage TNF-α responses during A fumigatus infection (Figure 1E-H). These observations indicate that Ibrutinib blocks inflammatory responses to A fumigatus in human macrophages through a Btk-dependent pathway.
![Figure 1. Ibrutinib blocks Btk-dependent activation of NFAT and NF-κB in human macrophages during A fumigatus infection. (A) A fumigatus induces autophosphorylation of Btk at Tyr 223, and this is inhibited by Ibrutinib. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (multiplicity of infection [MOI] = 5) for up to 2 hours. Whole cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by western blotting. Membranes were probed with anti-pBTK and anti-BTK antibodies. (B) BTK phosphorylation is required for NFAT and NF-κB activation in response to A fumigatus in THP1 macrophages. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (MOI = 5) for up to 2 hours. Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-NFATc1, NF-κB, and HDAC antibodies. (C-D) BTK mediates NFAT and NF-ĸB activation pathways in hMDMs (supplemental Figure 2). Monocyte-derived macrophages were pretreated with Scramble or BTK-targeting siRNA (75 nM) for 72 hours. Cells were stimulated with eGFP A fumigatus swollen conidia (MOI = 1) for 1 hour, and NFATc1 and NF-κB translocation were measured by confocal microscopy. Nuclear translocation was quantified by calculating the percent overlap of the nuclear DAPI and transcription factor-linked fluorophore channels. Data were calculated from 7 fields of view taken at random per biological repeat. Mean and standard deviation of 3 biological repeats are represented. Statistical analysis was performed using paired Student t tests: ns, not significant; *P < .05; NS, nonstimulated. N = 3. Af, A fumigatus; Ca, C albicans; Zym, zymosan. (E-H) TNF-α release by human macrophages in response to A fumigatus is BTK dependent. Monocyte-derived macrophages (E,G-H) and alveolar macrophages (F) were pretreated with Ibrutinib (1 µM) for 1 hour or scramble or BTK-targeting siRNA (100 nM) for 72 hours. (E-G) Macrophages were stimulated with A fumigatus swollen conidia (MOI = 1) for 16 hours. TNF-α levels in the tissue culture supernatants were measured by enzyme-linked immunosorbent assay. (H) Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-BTK and anti–β-actin antibodies. Statistical analysis was performed using paired Student t tests. *P < .05. NS, nonstimulated. N = 3 to 5.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/18/10.1182_blood-2017-12-823393/4/m_blood823393f1.png?Expires=1767697858&Signature=R5jC2JCafOytvqow-wtlyI3qid6nA-NLcRjLPtfUSONVEUEaP4c33HDg3JzujsWNBbvLRAPMelQoWT0-oEKXU17He~F-~TBr00R38XtQxZly2MD~V0oDaDCh-u2rDKxXN7nDYn1OZEEbEbmq4Y6JlzG2yEmm4KSQwD75NPUfmWtnfegxSpCHctiSzdMBggRn~RuFdI-HfDXfglp5iN2Avu4a1TT1CRUin-JPg6667wRmiNz-FTRtipLE0cnLFztHuiCKjXYzMpfahoLc8QS2YJAraejDl5~pxKlb3sGoZ2hVkVTYPapyWJ34pYlapCfDHfJJJzhlU-S3MAY6NROhqA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ibrutinib blocks Btk-dependent activation of NFAT and NF-κB in human macrophages during A fumigatus infection. (A) A fumigatus induces autophosphorylation of Btk at Tyr 223, and this is inhibited by Ibrutinib. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (multiplicity of infection [MOI] = 5) for up to 2 hours. Whole cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by western blotting. Membranes were probed with anti-pBTK and anti-BTK antibodies. (B) BTK phosphorylation is required for NFAT and NF-κB activation in response to A fumigatus in THP1 macrophages. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (MOI = 5) for up to 2 hours. Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-NFATc1, NF-κB, and HDAC antibodies. (C-D) BTK mediates NFAT and NF-ĸB activation pathways in hMDMs (supplemental Figure 2). Monocyte-derived macrophages were pretreated with Scramble or BTK-targeting siRNA (75 nM) for 72 hours. Cells were stimulated with eGFP A fumigatus swollen conidia (MOI = 1) for 1 hour, and NFATc1 and NF-κB translocation were measured by confocal microscopy. Nuclear translocation was quantified by calculating the percent overlap of the nuclear DAPI and transcription factor-linked fluorophore channels. Data were calculated from 7 fields of view taken at random per biological repeat. Mean and standard deviation of 3 biological repeats are represented. Statistical analysis was performed using paired Student t tests: ns, not significant; *P < .05; NS, nonstimulated. N = 3. Af, A fumigatus; Ca, C albicans; Zym, zymosan. (E-H) TNF-α release by human macrophages in response to A fumigatus is BTK dependent. Monocyte-derived macrophages (E,G-H) and alveolar macrophages (F) were pretreated with Ibrutinib (1 µM) for 1 hour or scramble or BTK-targeting siRNA (100 nM) for 72 hours. (E-G) Macrophages were stimulated with A fumigatus swollen conidia (MOI = 1) for 16 hours. TNF-α levels in the tissue culture supernatants were measured by enzyme-linked immunosorbent assay. (H) Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-BTK and anti–β-actin antibodies. Statistical analysis was performed using paired Student t tests. *P < .05. NS, nonstimulated. N = 3 to 5.
Ibrutinib blocks Btk-dependent activation of NFAT and NF-κB in human macrophages during A fumigatus infection. (A) A fumigatus induces autophosphorylation of Btk at Tyr 223, and this is inhibited by Ibrutinib. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (multiplicity of infection [MOI] = 5) for up to 2 hours. Whole cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by western blotting. Membranes were probed with anti-pBTK and anti-BTK antibodies. (B) BTK phosphorylation is required for NFAT and NF-κB activation in response to A fumigatus in THP1 macrophages. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (MOI = 5) for up to 2 hours. Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-NFATc1, NF-κB, and HDAC antibodies. (C-D) BTK mediates NFAT and NF-ĸB activation pathways in hMDMs (supplemental Figure 2). Monocyte-derived macrophages were pretreated with Scramble or BTK-targeting siRNA (75 nM) for 72 hours. Cells were stimulated with eGFP A fumigatus swollen conidia (MOI = 1) for 1 hour, and NFATc1 and NF-κB translocation were measured by confocal microscopy. Nuclear translocation was quantified by calculating the percent overlap of the nuclear DAPI and transcription factor-linked fluorophore channels. Data were calculated from 7 fields of view taken at random per biological repeat. Mean and standard deviation of 3 biological repeats are represented. Statistical analysis was performed using paired Student t tests: ns, not significant; *P < .05; NS, nonstimulated. N = 3. Af, A fumigatus; Ca, C albicans; Zym, zymosan. (E-H) TNF-α release by human macrophages in response to A fumigatus is BTK dependent. Monocyte-derived macrophages (E,G-H) and alveolar macrophages (F) were pretreated with Ibrutinib (1 µM) for 1 hour or scramble or BTK-targeting siRNA (100 nM) for 72 hours. (E-G) Macrophages were stimulated with A fumigatus swollen conidia (MOI = 1) for 16 hours. TNF-α levels in the tissue culture supernatants were measured by enzyme-linked immunosorbent assay. (H) Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-BTK and anti–β-actin antibodies. Statistical analysis was performed using paired Student t tests. *P < .05. NS, nonstimulated. N = 3 to 5.
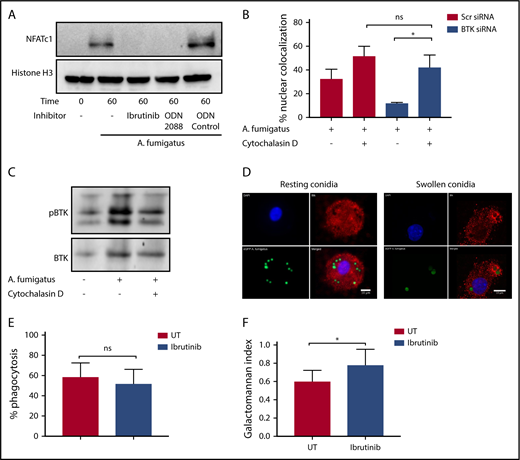
Our murine studies indicated that Btk-dependent macrophage responses to A fumigatus are mediated through an endosomal TLR9 signaling pathway.4 Consistent with these observations, A fumigatus–dependent NFAT translocation in hMDMs was blocked by both Ibrutinib and the TLR9-blocking nucleotide ODN2088 (Figure 2A). Furthermore, inhibition of phagocytosis of A fumigatus by hMDMs using cytochalasin D led to a loss of Btk-dependency for NFAT-dependent signaling responses (Figure 2B). In accordance with this finding, cytochalasin D inhibited Btk phosphorylation in hMDMs during A fumigatus infection (Figure 2C). Using confocal fluorescence microscopy, we conformed that Btk colocalizes with swollen, but not resting, A fumigatus conidia during phagocytosis (Figure 2D). However, Ibrutinib had no inhibitory effect on phagocytosis (Figure 2E). Ibrutinib impaired fungal growth control by macrophages (Figure 2F). These observations suggest that A fumigatus–dependent macrophage Btk signaling is endosomally driven and dependent on TLR9. Btk has also been shown to regulate reactive oxygen species production, inflammasome activation, chemotaxis, and adhesion in myeloid cells.6-10 Further studies have shown that BTK and TEC kinase are also important for myeloid immunity to C albicans.11,12 Furthermore, Ibrutinib is known to have off-target effects on other TEC kinases.13,14 Future studies should focus on defining the wider impact of Ibrutinib on TEC kinase-dependent innate immunity to fungi.15
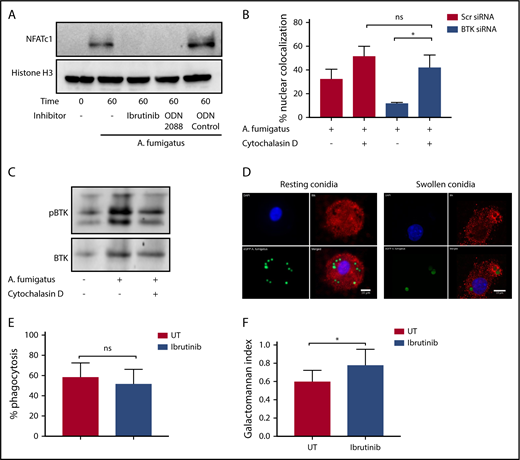
Endosomally driven Btk responses during human macrophage infection with A fumigatus are required for optimal fungal growth control. (A) TLR9 engagement and BTK phosphorylation are required for NFAT activation in response to A fumigatus in hMDMS. hMDMs were pretreated with Ibrutinib (1 µM), the TLR9-blocking nucleotide ODN2088 (10 µM), or ODN nucleotide control (10 µM) for 1 hour. Macrophages were then infected with A fumigatus swollen conidia (MOI = 1) for 1 hour. Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-NFATc1 and Histone H3 antibodies. (B-C) BTK mediates an endosomal NF-ĸB activation pathway in human macrophages. (B) Monocyte-derived macrophages were pretreated with Scramble or BTK-targeting siRNA (100 nM) for 72 hours and additionally with cytochalasin D (10 µM) or vehicle. Macrophages were stimulated with eGFP A fumigatus swollen conidia (MOI = 1) for 1 hour, and NFATc1 and NFKB translocation were measured by confocal microscopy. Nuclear translocation was quantified by calculating the percent overlap of the nuclear DAPI and transcription factor–linked fluorophore channels. Data were calculated from 7 fields of view taken at random per biological repeat. Mean and standard deviation of 3 biological repeats are represented. (C) Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-BTK and anti–β-actin antibodies. Statistical analysis was performed using paired Student t tests: ns, not significant; *P < .05; NS, nonstimulated. (D) BTK is recruited to the membranes of endosomes containing swollen but not resting conidia in hMDMs. Representative confocal microscopy of monocyte-derived macrophages infected with eGFP expressing A fumigatus (green) (MOI = 1) at 45 minutes propidium iodide stained for BTK (red) and nuclei (blue) (original magnification ×60). (a) Nuclei; (b) BTK; (c) eGFP; (d) composite images. (E) Ibrutinib does not block A fumigatus phagocytosis. Monocyte-derived macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Macrophages were infected with biotinylated A fumigatus-GFP (green fluorescent protein) swollen conidia (MOI = 1) for 2 hours. External conidia were then counterstained with Cy3 biotin antibody, and phagocytosis was measured by flow cytometry. (F) Ibrutinib impairs macrophage control of fungal growth in vitro. Monocyte-derived macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (MOI = 1) for 6 hours. Galactomannan levels were measured in the tissue culture supernatants. UT, untreated.
Endosomally driven Btk responses during human macrophage infection with A fumigatus are required for optimal fungal growth control. (A) TLR9 engagement and BTK phosphorylation are required for NFAT activation in response to A fumigatus in hMDMS. hMDMs were pretreated with Ibrutinib (1 µM), the TLR9-blocking nucleotide ODN2088 (10 µM), or ODN nucleotide control (10 µM) for 1 hour. Macrophages were then infected with A fumigatus swollen conidia (MOI = 1) for 1 hour. Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-NFATc1 and Histone H3 antibodies. (B-C) BTK mediates an endosomal NF-ĸB activation pathway in human macrophages. (B) Monocyte-derived macrophages were pretreated with Scramble or BTK-targeting siRNA (100 nM) for 72 hours and additionally with cytochalasin D (10 µM) or vehicle. Macrophages were stimulated with eGFP A fumigatus swollen conidia (MOI = 1) for 1 hour, and NFATc1 and NFKB translocation were measured by confocal microscopy. Nuclear translocation was quantified by calculating the percent overlap of the nuclear DAPI and transcription factor–linked fluorophore channels. Data were calculated from 7 fields of view taken at random per biological repeat. Mean and standard deviation of 3 biological repeats are represented. (C) Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-BTK and anti–β-actin antibodies. Statistical analysis was performed using paired Student t tests: ns, not significant; *P < .05; NS, nonstimulated. (D) BTK is recruited to the membranes of endosomes containing swollen but not resting conidia in hMDMs. Representative confocal microscopy of monocyte-derived macrophages infected with eGFP expressing A fumigatus (green) (MOI = 1) at 45 minutes propidium iodide stained for BTK (red) and nuclei (blue) (original magnification ×60). (a) Nuclei; (b) BTK; (c) eGFP; (d) composite images. (E) Ibrutinib does not block A fumigatus phagocytosis. Monocyte-derived macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Macrophages were infected with biotinylated A fumigatus-GFP (green fluorescent protein) swollen conidia (MOI = 1) for 2 hours. External conidia were then counterstained with Cy3 biotin antibody, and phagocytosis was measured by flow cytometry. (F) Ibrutinib impairs macrophage control of fungal growth in vitro. Monocyte-derived macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (MOI = 1) for 6 hours. Galactomannan levels were measured in the tissue culture supernatants. UT, untreated.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by the Wellcome Trust Strategic Award in Medical Mycology and Fungal Immunology (G097377) (A.B., A.W.), by an MRC Clinical Research Fellowship (MR/K002708/1) (A.S.), by the MRC Centre for Medical Mycology (MR/N006364/1) at the University of Aberdeen (A.W.), and by a Wellcome Trust Seed Award (204566/Z/16/Z) (D.A.-J.).
Authorship
Contribution: D.A.-J., A.W., and A.B. conceived the study; A.B., T.C., and A.S. performed the experiments; and A.B., T.C., A.S., A.W., and D.A.-J. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Darius Armstrong-James, Fungal Pathogens Laboratory, Sir Alexander Fleming Building, National Heart and Lung Institute, Imperial College London, SWZ 2AZ, London, United Kingdom; e-mail: d.armstrong@imperial.ac.uk.

![Figure 1. Ibrutinib blocks Btk-dependent activation of NFAT and NF-κB in human macrophages during A fumigatus infection. (A) A fumigatus induces autophosphorylation of Btk at Tyr 223, and this is inhibited by Ibrutinib. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (multiplicity of infection [MOI] = 5) for up to 2 hours. Whole cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by western blotting. Membranes were probed with anti-pBTK and anti-BTK antibodies. (B) BTK phosphorylation is required for NFAT and NF-κB activation in response to A fumigatus in THP1 macrophages. THP1 macrophages were pretreated with Ibrutinib (1 µM) for 1 hour. Cells were stimulated with A fumigatus swollen conidia (MOI = 5) for up to 2 hours. Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-NFATc1, NF-κB, and HDAC antibodies. (C-D) BTK mediates NFAT and NF-ĸB activation pathways in hMDMs (supplemental Figure 2). Monocyte-derived macrophages were pretreated with Scramble or BTK-targeting siRNA (75 nM) for 72 hours. Cells were stimulated with eGFP A fumigatus swollen conidia (MOI = 1) for 1 hour, and NFATc1 and NF-κB translocation were measured by confocal microscopy. Nuclear translocation was quantified by calculating the percent overlap of the nuclear DAPI and transcription factor-linked fluorophore channels. Data were calculated from 7 fields of view taken at random per biological repeat. Mean and standard deviation of 3 biological repeats are represented. Statistical analysis was performed using paired Student t tests: ns, not significant; *P < .05; NS, nonstimulated. N = 3. Af, A fumigatus; Ca, C albicans; Zym, zymosan. (E-H) TNF-α release by human macrophages in response to A fumigatus is BTK dependent. Monocyte-derived macrophages (E,G-H) and alveolar macrophages (F) were pretreated with Ibrutinib (1 µM) for 1 hour or scramble or BTK-targeting siRNA (100 nM) for 72 hours. (E-G) Macrophages were stimulated with A fumigatus swollen conidia (MOI = 1) for 16 hours. TNF-α levels in the tissue culture supernatants were measured by enzyme-linked immunosorbent assay. (H) Whole cell lysates were separated by SDS-PAGE, followed by western blotting. Membranes were probed with anti-BTK and anti–β-actin antibodies. Statistical analysis was performed using paired Student t tests. *P < .05. NS, nonstimulated. N = 3 to 5.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/18/10.1182_blood-2017-12-823393/4/m_blood823393f1.png?Expires=1767697859&Signature=MNHvjxN-CEfKDTRJKMcWH5M1TdeE3A2R-yaLZly1gl3ReFYtmXrv1nHyW2Lrd7aAspj4nVzuHfZdMVlhbcursxyQhQx3VI8w~p5ZOL2Af8FZMp35TBkm-hVqV~g9VImDh7SUPseV8KGGLEaDs1uj8hNt9H1abPqB9V0Px8qV5ova0D2gTl0cnaqRPankI8O40g6y~3JCk554N1Dj2fqC9eI45W1IwEUG8ANXb02d~X9A5MydJff4PhS7uQjehDBUD~S00LPrbJTn0M3VsgiSpuDs4SUHJ82sHLi~eAsdgaOTEaHGs7V9tPGkqRHdl~67c~M~WzmYqjlxzol1BXE7vg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)