

Abstract
Introduction: Acalabrutinib is a Bruton tyrosine kinase (BTK) inhibitor approved for treatment of relapsed patients (pts) with mantle cell lymphoma. We have reported previously that ibrutinib refractory MCL pts have poor survival. However, outcomes, causes of discontinuation, management and the genomic landscape of MCL in pts who discontinued acalabrutinib are rarely reported.
Method: We reviewed charts from all MCL pts treated with single agent acalabrutinib (n=28) in the relapsed setting and identified 15 pts who discontinued acalabrutinib and who are described in this analysis. Outcome after discontinuing acalabrutinib is reported. Whole-exome sequencing (WES) with SureSelect Human All Exon V6 was performed on 10 tumor specimens and 5 matched germline samples collected from 9 pts whose MCL progressed on acalabrutinib; among these pts 4 tumors were collected at baseline and 6 were collected after disease progression. One patient had sufficient DNAs available for both time points (baseline and progression).
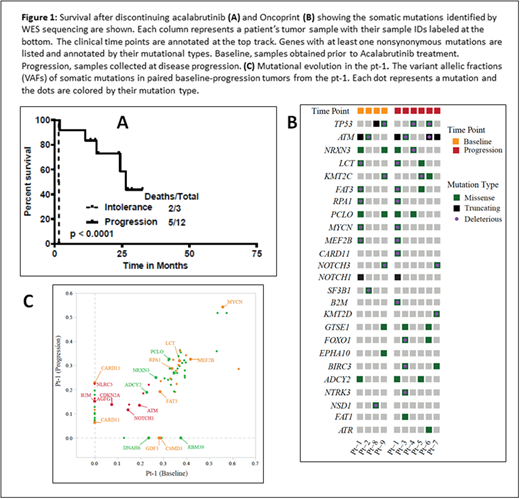
Results: The median duration on treatment with acalabrutinib was 6.5 months (1 to 29 months) and the median number of cycles of acalabrutinib treatment was 6 (range, 1-30). Seven pts had complete remission (CR) as their best response on acalabrutinib, 5 were primary refractory and 3 achieved partial remission. In 12 pts (80%) acalabrutinib was discontinued due to disease progression (2 pts transformed from classic to blastoid and pleomorphic type at progression) and 3 pts were discontinued due to intolerance (one for fatigue and idiopathic encephalopathy, one due to unrelated severe aortic stenosis and another for cytopenias secondary to therapy related myelodysplasia; all three pts were in CR). Nine pts had classic and 3 pts each had blastoid or pleomorphic features before starting acalabrutinib. Overall, median Ki-67 expression was 50% (range, 5-100) and all pts had high a MIPI score. The median number of prior treatments was 1 (range, 1-3); all chemo-immunotherapy (10 pts were previously treated with rituximab-hyper-CVAD) and none with ibrutinib. Two pts who transformed on acalabrutinib received acalabrutinib for a median duration of 12 months (range, 8-16.5). Median follow up after discontinuation was 27 months and the median survival was 25 months (26 months for progression and 1.5 months for intolerance; p <0.001, Figure-1A). Patients who discontinued due to intolerance did not get subsequent treatment for MCL. Among the 12 pts who progressed on acalabrutinib, 11 pts received systemic therapy for MCL [seven received ibrutinib based therapies (2 non responders, 3 achieved CR and 2 were PR and all pts progressed subsequently), 3 got chemo-immunotherapy and progressed and one pt did not receive any treatment and was lost to follow up and died. Six patients received a clinical trial with CAR-T cells (results will be reported separately). Overall, at the time of last follow up, 8 pts were alive and 7 were in CR.
Recurrently mutated genes in these tumors included ATM (6/10; 60%), TP53 (4/10; 40%), KMT2C (3/10), MYCN (2/10), NOTCH1 (2/10), NOTCH3 (2/10), and MEF2B (2/10) (Fig. 1B). We did not detect any mutation or copy number alterations in BTK, PLCG2, TRAF2/3 and MYD88 that have been reported previously to be associated with ibrutinib resistance. Compared to tumors at baseline, ATM was mutated at a higher frequency in samples at progression (67% vs. 50%; p=NS). To investigate the mutation evolution on acalabrutinib treatment, mutation profiles, particularly the mutation variant allelic fractions (VAFs), were compared between the baseline and progression samples from pt-1 (Fig. 1C). Mutation of MYCN, MEF2B, ATM, and NOTCH1 were identified in both tumors at similar VAFs, whereas mutation of CARD11 (two mutations), NLRC5 and B2M were detected only at progression. In pt-1, both the NLRC5 and β2M mutations acquired at disease progression were truncating, suggesting loss-of-function alterations. Copy number analysis reveals frequent whole-genome doubling and intensive copy number alterations in all tumors, including recurrent losses of chromosome 9p, 17p, and chromosome 13, indicating chromosomal instability as a driver of disease progression.
Conclusion: Patients who progress on acalabrutinib have a poor outcome, and newer therapies are required for their treatment. In this small cohort, we observed non-BTK mutations associated with acalabrutinib resistance and disease progression.
Nastoupil:Genentech: Honoraria, Research Funding; TG Therappeutics: Research Funding; Spectrum: Honoraria; Gilead: Honoraria; Merck: Honoraria, Research Funding; Janssen: Research Funding; Celgene: Honoraria, Research Funding; Karus: Research Funding; Novartis: Honoraria; Juno: Honoraria. Neelapu:Celgene: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding; Cellectis: Research Funding; Poseida: Research Funding; Merck: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding; Acerta: Research Funding; Karus: Research Funding; Bristol-Myers Squibb: Research Funding; Unum Therapeutics: Membership on an entity's Board of Directors or advisory committees; Kite/Gilead: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding; Novartis: Membership on an entity's Board of Directors or advisory committees. Fowler:Janssen: Consultancy, Research Funding; Pharmacyclics: Consultancy, Research Funding. Wang:Acerta Pharma: Honoraria, Research Funding; MoreHealth: Consultancy; AstraZeneca: Consultancy, Research Funding; Kite Pharma: Research Funding; Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Dava Oncology: Honoraria; Juno: Research Funding; Pharmacyclics: Honoraria, Research Funding; Janssen: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Novartis: Research Funding.

