

Key Points
MHC II cell-autonomously regulates self-renewal and differentiation in developing B-cell precursors.
MHC II expression restrains growth of B-cell leukemias in vitro and in vivo, independent of CD4+ T-cell surveillance.

Abstract
Best known for presenting antigenic peptides to CD4+ T cells, major histocompatibility complex class II (MHC II) also transmits or may modify intracellular signals. Here, we show that MHC II cell-autonomously regulates the balance between self-renewal and differentiation in B-cell precursors, as well as in malignant B cells. Initiation of MHC II expression early during bone marrow B-cell development limited the occupancy of cycling compartments by promoting differentiation, thus regulating the numerical output of B cells. MHC II deficiency preserved stem cell characteristics in developing pro-B cells in vivo, and ectopic MHC II expression accelerated hematopoietic stem cell differentiation in vitro. Moreover, MHC II expression restrained growth of murine B-cell leukemia cell lines in vitro and in vivo, independently of CD4+ T-cell surveillance. Our results highlight an important cell-intrinsic contribution of MHC II expression to establishing the differentiated B-cell phenotype.
Introduction
Growth and differentiation of healthy and malignant B cells is regulated by cell-intrinsic processes, as well as cellular communication, such as B-cell receptor (BCR)-derived signals,1 and interaction with CD4+ T cells, recognizing B-cell–presented peptide-major histocompatibility complex class II (MHC II) complexes.2 MHC II recognition by CD4+ T cells can also lead to killing of target B cells, contributing to the elimination of infected or transformed cells3,4 and selecting for the loss of MHC II during viral infection or cancer evolution.5,6
However, the MHC II α and β chains additionally demonstrate signaling capacity, with their in vitro engagement by the T-cell receptor, bacterial superantigens, or crosslinking antibodies, triggering multiple signaling cascades.7-11 Indeed, ligation of MHC II molecules has long been recognized as a potent inducer of in vitro B-cell proliferation and differentiation,12,13 as well as homotypic adhesion, cytokine production, or apoptosis.8,10 Moreover, antibody ligation of human MHC molecules induces programmed cell death of malignant lymphoid cells in vitro and in vivo.14
The diversity of possible outcomes that have been described to follow MHC II crosslinking is likely explained by context-dependent association of MHC II chains with an extended array of signal transducers or adaptors,7-11 or by MHC II–mediated alteration of signaling cascades initiated by other receptors. For instance, a role for MHC II in modulating immune cell responsiveness to acute stimuli, such as lipopolysaccharide and other microbial products, has long been recognized, although opposite effects in B cells and myeloid cells are reported.15-19
These observations suggest a possible role for MHC II in immune cell physiology or pathology. Despite recognition of the cell-intrinsic effect of in vitro MHC II crosslinking in B-cell differentiation over 3 decades ago,12,13,20 the contribution of cell-autonomous MHC II signaling to immune cell development, function, or pathology in vivo remains to be understood. Here, we reveal a cell-autonomous role for MHC II expression in determining the balance between self-renewal and differentiation in B-cell precursors and in malignant B cells.
Methods
Additional methods are available in supplemental Methods (available on the Blood Web site).
Mice
Inbred C57BL/6J (B6) and CD45.1+ congenic B6 (B6.SJL-Ptprca Pep3b/BoyJ) mice were originally obtained from The Jackson Laboratory. Mice carrying a conditional H2-Ab1 allele (H2-Ab1c)21 were crossed to mice expressing Cre in dendritic cells (DCs) under the Itgax (CD11c-Cre driver; Itgaxcre)22 or the Zbtb46 promoter (zDC-Cre driver; Zbtb46cre),23 or in early and late B-cell lineages, under the Cd79a (mb1-Cre; Cd79acre)24 and the Fcer2a (CD23-Cre; Fcer2acre)25 promoter, respectively. Due to frequent germline deletion of the conditional H2-Ab1 allele inherited from the Cre−-transmitting parent, H2-Ab1c mice contained only 1 functional conditional allele, inherited from the Cre− parent, and therefore expressed reduced levels of MHC II in Cre− cells. Mice constitutively lacking all conventional MHC II genes (H2dlAb1-Ea),26 Rag1-deficient (Rag1−/−) mice,27 and Rag2-deficient (Rag2−/−) mice28 have been previously described. All mouse strains were on the B6 genetic background and maintained at the Francis Crick Institute’s animal facilities. All animal experiments were approved by the ethical committee of the Francis Crick Institute, and conducted according to local guidelines and UK Home Office regulations under the Animals Scientific Procedures Act 1986 (ASPA).
Competitive bone marrow chimeras
Bone marrow cell suspensions were prepared by flushing the bone cavities of femurs and tibiae from donor mice with air-buffered Iscove’s modified Dulbecco’s medium. In certain experiments, T cells were depleted from bone marrow cell suspensions by immunomagnetic negative selection, using the EasySep system (StemCell Technologies). Recipient mice were preconditioned by intraperitoneal injection of the myeloablative drug Busilvex (Pierre Fabre Ltd), at a dose of 10 mg/kg of body weight, dissolved in phosphate-buffered saline. They then received an IV injection of donor bone marrow cell mixtures (a total of 107 cells in Iscove's modified Dulbecco's medium), 24 hours after myeloablation. The degree of reconstitution was assessed in recipient mice 8 to 10 weeks post–bone marrow transplantation.
Flow cytometry and cell sorting
Single-cell suspensions were stained for 20 minutes at room temperature or at 4°C with directly conjugated antibodies to surface antigens (listed in supplemental Table 1). Immune cell subtypes were identified using gating strategies depicted in supplemental Figure 1. Multicolor cytometry data were acquired with LSRFortessa X-20 (BD Biosciences) flow cytometers, running BD FACSDiva v8.0, and analyzed with FlowJo v10 (TreeStar Inc) analysis software. Cell populations of interest were purified (>98% purity) by cell sorting, performed on a FACSAria Fusion flow cytometer (BD Biosciences) or MoFlo cell sorters (DakoCytomation).
Large-scale fluorescence microscopy
Pro-B cells were enriched by cell sorting from wild-type (WT) or H2dlAb1-Ea bone marrow samples and were then used for large-scale fluorescence image collection and analysis as previously described.29
Statistical analyses
Statistical comparisons were made using SigmaPlot 13 (Systat Software Inc). Parametric comparisons of normally distributed values that satisfied the variance criteria were made by unpaired Student t tests or 1-way analysis of variances (ANOVAs). Data that did not pass the variance test were compared with the nonparametric 2-tailed Mann-Whitney rank-sum test or ANOVA-on-ranks tests. Calculation of correlation coefficients was performed using Excel 2016. Analysis of processed RNA sequencing (RNAseq) data, hierarchical clustering, and heat-map production was performed with Omics Explorer 3.3 (Qlucore, Lund, Sweden).
Results
Initiation of MHC II expression shapes B-cell development potential
To investigate the cell-autonomous functions of MHC II, we used Cre-mediated ablation of a conditional H2-Ab1 allele (H2-Ab1c) encoding the MHC II Aβ chain. Here, incomplete and ectopic Cre activity in on-target and off-target cell types, respectively, creates a degree of H2-Ab1 deletion mosaicism, permitting comparison between MHC II–expressing and MHC II–deleted cells within the same host.
DC-targeted Cre expression (CD11c-Cre driver; Itgaxcre) expectedly abolished MHC II expression in not only DCs, but also surprisingly in the majority of B cells (Figure 1). Unexpectedly high proportions of B cells also lost MHC II expression when the more specific zDC-Cre driver (Zbtb46cre) was used (Figure 1). Furthermore, targeting Cre expression to early or immature B-cell lineages using the mb1-Cre (Cd79acre) or the CD23-Cre (Fcer2acre) driver, respectively, abolished MHC II expression in B cells, as well as in a considerable proportion of DCs (Figure 1).
![Figure 1. Overrepresentation of MHC II−cells in H2-Ab1c mice. (A) Flow cytometric identification of splenic B cells and DCs. (B-C) MHC II expression (B) and frequency (plus or minus standard error mean [SEM]) of MHC II− cells (C) in gated splenic DCs and B cells from mice of the indicated genotype. Symbols represent individual mice. *P < .001 between WT and all other genotypes by 1-way ANOVA.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/10/10.1182_blood-2018-11-885467/3/m_blood885467f1.png?Expires=1763749670&Signature=w-jdG0zBejTlYcgiqbexzeDNbBA54Tgiu35rvnXmunwt9NYFdcc1VUIOKhskAsOHMRlkuQNYr8ldf0TCztK9Fg-eaKn8YF6BW1As6d4McWnrHvs3apYSLqofzWrfTQnntYj6HLtlAKBktauUyurufGMRqdShDzVd2-01kniWU-utbI1zV3oRfcksIgH6feaZo-4tXFnDGlN3B23lf93A-H-9c7Qxmb8yMeuEJuoTvx9MtRhOcc1Tg9IjBgr2vI4-uQHcwSKRDhCKGv9VwGBfJJROiFqNFxNfqY4TACOsLJZN-iLtANhswZvqP5SXHURz87dCmaWJP5MhKCW66n9jRA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overrepresentation of MHC II−cells in H2-Ab1cmice. (A) Flow cytometric identification of splenic B cells and DCs. (B-C) MHC II expression (B) and frequency (plus or minus standard error mean [SEM]) of MHC II− cells (C) in gated splenic DCs and B cells from mice of the indicated genotype. Symbols represent individual mice. *P < .001 between WT and all other genotypes by 1-way ANOVA.
Overrepresentation of MHC II−cells in H2-Ab1cmice. (A) Flow cytometric identification of splenic B cells and DCs. (B-C) MHC II expression (B) and frequency (plus or minus standard error mean [SEM]) of MHC II− cells (C) in gated splenic DCs and B cells from mice of the indicated genotype. Symbols represent individual mice. *P < .001 between WT and all other genotypes by 1-way ANOVA.
Although it was possible that off-target loss of MHC II was caused by excessive ectopic Cre-mediated recombination, this was inconsistent with the reported activities of the Cre drivers used.22-25 We therefore considered the alternative hypothesis that the unexpectedly high frequency of apparent off-target loss of MHC II in the Cre drivers tested was accentuated by aberrant lineage commitment or selective outgrowth of initially rare MHC II–deleted cells. To probe this possibility and to circumvent the inflammatory or myeloproliferative disease that develops in ItgaxcreH2-Ab1c mice,23,30 we set up competitive bone marrow chimeras between MHC II–deficient and WT mice (Figure 2A-E). Interestingly, bone marrow from ItgaxcreH2-Ab1c or Fcer2acreH2-Ab1c donors produced preferentially B cells, at the expense of T cells and myeloid cells (Figure 2A-D). Consequently, several subsets of mature B cells were significantly enriched in MHC II–deleted B cells, with the exception of germinal center (GC) B cells (Figure 2E-G), which were exclusively MHC II+, due to their dependency on CD4+ T-cell help for survival.31 Comparable results were obtained when H2dlAb1-Ea mice, constitutively deleted in all MHC II–encoding genes, were used as donors (supplemental Figure 2A-B), arguing against a secondary effect of Cre-mediated deletion or of expression of unpaired MHC class II chains.32

Competitive advantage of MHC II−B cells. (A) Competitive bone marrow chimeras were setup by reconstituting myeloablated CD45.1+CD45.2+ WT (WT) hosts with mixtures of CD45.1+ control (donor 1; always WT) and CD45.2+ test genotype (donor 2; WT, ItgaxcreH2-Ab1c, or Fcer2acreH2-Ab1c) bone marrow. The composition of myeloid and lymphoid cells in reconstituted recipients was then examined by flow cytometry (the key refers to panels B-D). (B-D) B220 and MHC II expression and mean percentage of splenic T cells, B cells, and myeloid cells originating from the host or each of the 2 types of donor, in competitive chimeras between WT and WT donors (n = 5) (B); WT and ItgaxcreH2-Ab1c donors (n = 8) (C); or WT and Fcer2acreH2-Ab1c donors (n = 7) (D). (E) Gating of splenic transitional (T) T1, T2, T3, marginal zone (MZ), follicular (FO), and GC B cells. (F) Mean change (plus or minus SEM) from input frequency of CD45.2+ donor cells in splenic T cells or T1, T2, T3, MZ, FO, and GC B cells from chimeras between CD45.1+ WT control and CD45.2+ test genotype donors (WT: WT, n = 12; WT: ItgaxcreH2-Ab1c, n = 6; WT: Fcer2acreH2-Ab1c, n = 5). *P < .01; **P < .001 between WT and other genotypes by Student t tests. (G) MHC II expression in splenic FO and overlaid GL7+ GC B cells of host and donor origin, distinguished by CD45 allotype expression. FACS, fluorescence-activated cell sorting.
Competitive advantage of MHC II−B cells. (A) Competitive bone marrow chimeras were setup by reconstituting myeloablated CD45.1+CD45.2+ WT (WT) hosts with mixtures of CD45.1+ control (donor 1; always WT) and CD45.2+ test genotype (donor 2; WT, ItgaxcreH2-Ab1c, or Fcer2acreH2-Ab1c) bone marrow. The composition of myeloid and lymphoid cells in reconstituted recipients was then examined by flow cytometry (the key refers to panels B-D). (B-D) B220 and MHC II expression and mean percentage of splenic T cells, B cells, and myeloid cells originating from the host or each of the 2 types of donor, in competitive chimeras between WT and WT donors (n = 5) (B); WT and ItgaxcreH2-Ab1c donors (n = 8) (C); or WT and Fcer2acreH2-Ab1c donors (n = 7) (D). (E) Gating of splenic transitional (T) T1, T2, T3, marginal zone (MZ), follicular (FO), and GC B cells. (F) Mean change (plus or minus SEM) from input frequency of CD45.2+ donor cells in splenic T cells or T1, T2, T3, MZ, FO, and GC B cells from chimeras between CD45.1+ WT control and CD45.2+ test genotype donors (WT: WT, n = 12; WT: ItgaxcreH2-Ab1c, n = 6; WT: Fcer2acreH2-Ab1c, n = 5). *P < .01; **P < .001 between WT and other genotypes by Student t tests. (G) MHC II expression in splenic FO and overlaid GL7+ GC B cells of host and donor origin, distinguished by CD45 allotype expression. FACS, fluorescence-activated cell sorting.
Next, we explored when during B-cell development MHC II–deleted cells might have an advantage over WT competitors and whether MHC II expression was already present at that developmental stage. We adopted Hardy fraction (Fr) nomenclature,33 which subdivides bone marrow B cells into distinct stages of progressive differentiation. Fr A cells represent a heterogeneous population of the least differentiated B-cell precursors, whereas Fr B and C cells are committed, highly proliferative pro-B and pre-B cells. Fr D cells typically exit the extensive cell division of the earlier Fr B and C cells, and become quiescent, while continuing BCR rearrangement.1,34 Lastly, Fr E cells represent immature cells that express surface immunoglobulin M (IgM), whereas Fr F cells are mature recirculating B cells. Surface expression of MHC II in WT cells was first detectable by flow cytometry in Fr A cells, gradually increasing both in terms of frequency and intensity in subsequent Fr B to F (Figure 3A-C), in keeping with prior reports.35,36 By large-scale fluorescence microscopy, MHC II proteins were detected at the plasma membrane weakly in WT Fr A cells and more prominently in Fr B and Fr C cells (Figure 3D), further confirming MHC II expression within these early developmental fractions. Coinciding with the initiation of MHC II expression, the overrepresentation of MHC II–deficient B cells in mixed bone marrow chimeras manifested between Fr A and C (Figure 3E; supplemental Figure 3). In contrast, MHC II deficiency did not affect the starting frequency of hematopoietic stem cells (HSCs) with in vitro B-cell potential (supplemental Figure 4). Collectively, these findings suggest that MHC II affects progression through the B-cell developmental stage at which it is first expressed (Fr A to C).
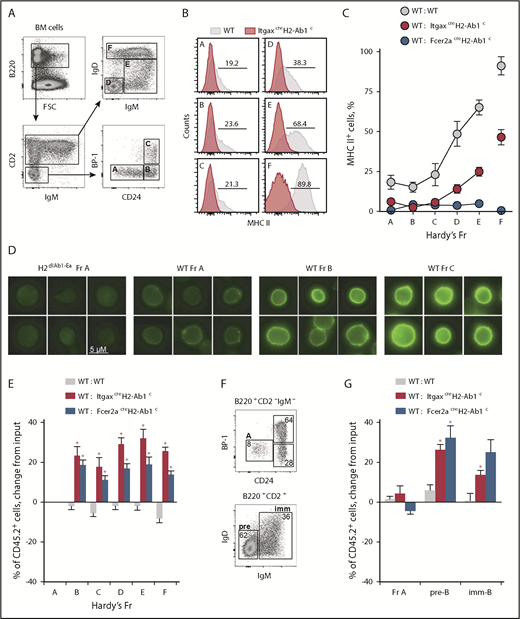
Initiation of MHC II expression shapes B-cell development. (A) Flow cytometric identification of bone marrow Fr A-F B cells. (B-C) MHC II expression (B) and mean frequency (plus or minus SEM) of MHC II+ cells (C) in Fr A-F bone marrow B cells from mice of the indicated genotype (WT, n = 3; ItgaxcreH2-Ab1c, n = 5; Fcer2acreH2-Ab1c, n = 6). (D) MHC II expression (green), detected by immunofluorescence staining in WT Fr A-C bone marrow B cells (MHC II antibody clone M5/114.15.2 stain; scale bar, 5 μM). ItgaxcreH2-Ab1c Fr A bone marrow B cells are included as staining control. (E) Mean change (plus or minus SEM) from input frequency of CD45.2+ cells in Fr A-F bone marrow B cells from chimeras between CD45.1+ WT control and CD45.2+ test genotype donors (WT: WT, n = 9; WT: ItgaxcreH2-Ab1c, n = 6; WT: Fcer2acreH2-Ab1c, n = 5). *P < .001 between WT and other genotypes by Student t tests. (F-G) In vitro development of purified Fr A ItgaxcreH2-Ab1c B-cell precursors into pre-B (pre; B220+CD2+IgD−IgM−) and immature B cells (imm; B220+CD2+IgD−IgM+) (F), and mean change (plus or minus SEM) from input frequency of CD45.2+ cells in Fr A, pre-B cells, and immature B cells following in vitro development of purified Fr A cells in competitive cultures between CD45.1+ WT control and CD45.2+ test genotype input Fr A cells (G) (WT: WT, n = 6; WT: ItgaxcreH2-Ab1c, n = 2; WT: Fcer2acreH2-Ab1c, n = 8). *P < .02 between WT and other genotypes by Student t tests. FSC, forward scatter.
Initiation of MHC II expression shapes B-cell development. (A) Flow cytometric identification of bone marrow Fr A-F B cells. (B-C) MHC II expression (B) and mean frequency (plus or minus SEM) of MHC II+ cells (C) in Fr A-F bone marrow B cells from mice of the indicated genotype (WT, n = 3; ItgaxcreH2-Ab1c, n = 5; Fcer2acreH2-Ab1c, n = 6). (D) MHC II expression (green), detected by immunofluorescence staining in WT Fr A-C bone marrow B cells (MHC II antibody clone M5/114.15.2 stain; scale bar, 5 μM). ItgaxcreH2-Ab1c Fr A bone marrow B cells are included as staining control. (E) Mean change (plus or minus SEM) from input frequency of CD45.2+ cells in Fr A-F bone marrow B cells from chimeras between CD45.1+ WT control and CD45.2+ test genotype donors (WT: WT, n = 9; WT: ItgaxcreH2-Ab1c, n = 6; WT: Fcer2acreH2-Ab1c, n = 5). *P < .001 between WT and other genotypes by Student t tests. (F-G) In vitro development of purified Fr A ItgaxcreH2-Ab1c B-cell precursors into pre-B (pre; B220+CD2+IgD−IgM−) and immature B cells (imm; B220+CD2+IgD−IgM+) (F), and mean change (plus or minus SEM) from input frequency of CD45.2+ cells in Fr A, pre-B cells, and immature B cells following in vitro development of purified Fr A cells in competitive cultures between CD45.1+ WT control and CD45.2+ test genotype input Fr A cells (G) (WT: WT, n = 6; WT: ItgaxcreH2-Ab1c, n = 2; WT: Fcer2acreH2-Ab1c, n = 8). *P < .02 between WT and other genotypes by Student t tests. FSC, forward scatter.
MHC II deficiency delays B-cell development progression
Low-level MHC II expression in developing B cells is considered sufficient to mediate antigen presentation to T cells, albeit with reduced efficiency.37 To begin to investigate how MHC II expression might be influencing B-cell development, we first excluded a possible role for CD4+ T cells bearing MHC II–restricted T-cell receptors. Indeed, H2-Ab1c–deleted progenitors still outcompeted WT counterparts in mixed bone marrow chimeras generated in Rag1−/−H2dlAb1-Ea recipients, where CD4+ T-cell development is precluded (supplemental Figure 5A-D). Furthermore, competitive in vitro differentiation assays revealed that Fr A cells purified from ItgaxcreH2-Ab1c or Fcer2acreH2-Ab1c donors numerically outcompeted those from WT donors in subsequent developmental stages (Figure 3F-G).
In light of these findings, we reasoned that numbers of developing B cells might not be simply extrinsically regulated based on their expression of MHC II. Instead, MHC II expression and associated interaction with putative MHC II ligands might induce transcriptional changes intrinsic to developing B cells that would not be present in MHC II–deficient counterparts. This was directly investigated by transcriptional profiling of WT and ItgaxcreH2-Ab1c Fr D pre-B cells (Figure 4A,C). Within WT Fr D pre-B cells, expression of surface MHC II proteins marked more advanced maturation, as indicated by the expression of immunoglobulin genes, multiple MHC II–related genes, and other maturation markers (eg, Ccr6, Fcrl5, and Il9r) (Figure 4A; supplemental Table 2), further supported by comparison with splenic B cells (supplemental Figure 6A-B), as well as by publicly available expression data38 (Figure 4B).

MHC II deficiency delays B-cell development progression. (A) Expression of differentially expressed genes (at least twofold, P ≤ .05) between MHC II− and MHC II+ Fr D WT B-cell precursor samples (left) and of selected MHC II, immunoglobulin, and B-cell differentiation genes in MHC II− or MHC II+ WT and ItgaxcreH2-Ab1c Fr D cells (right). (B) Expression level of selected B-cell differentiation genes in the indicated stages of B-cell development. (C) Expression of differentially expressed genes (at least twofold, P ≤ .05, q = 0.05) between WT and ItgaxcreH2-Ab1c Fr D B-cell precursor samples (top) and of the top 7 genes overrepresented in ItgaxcreH2-Ab1c Fr D cells (bottom). WT and ItgaxcreH2-Ab1c FO B cells are also included for comparison. (D) Expression level of selected B-cell stem cell genes in the indicated stages of B-cell development. (E) Correlation of the transcriptional signature of ItgaxcreH2-Ab1c Fr D cells with those of CLP and Fr A cells. Data in panels B and C, and the comparative data in panel D were obtained from ImmGen. (F) Functional pathways revealed by enrichment analysis on genes differentially upregulated (at least twofold, P ≤ .05, q = 0.05) in ItgaxcreH2-Ab1c Fr D cells compared with WT Fr D cells.
MHC II deficiency delays B-cell development progression. (A) Expression of differentially expressed genes (at least twofold, P ≤ .05) between MHC II− and MHC II+ Fr D WT B-cell precursor samples (left) and of selected MHC II, immunoglobulin, and B-cell differentiation genes in MHC II− or MHC II+ WT and ItgaxcreH2-Ab1c Fr D cells (right). (B) Expression level of selected B-cell differentiation genes in the indicated stages of B-cell development. (C) Expression of differentially expressed genes (at least twofold, P ≤ .05, q = 0.05) between WT and ItgaxcreH2-Ab1c Fr D B-cell precursor samples (top) and of the top 7 genes overrepresented in ItgaxcreH2-Ab1c Fr D cells (bottom). WT and ItgaxcreH2-Ab1c FO B cells are also included for comparison. (D) Expression level of selected B-cell stem cell genes in the indicated stages of B-cell development. (E) Correlation of the transcriptional signature of ItgaxcreH2-Ab1c Fr D cells with those of CLP and Fr A cells. Data in panels B and C, and the comparative data in panel D were obtained from ImmGen. (F) Functional pathways revealed by enrichment analysis on genes differentially upregulated (at least twofold, P ≤ .05, q = 0.05) in ItgaxcreH2-Ab1c Fr D cells compared with WT Fr D cells.
Given the heterogeneity of WT Fr D pre-B cells, we then compared each of the 2 subsets distinguished by MHC II expression, with those from ItgaxcreH2-Ab1c Fr D pre-B cells. MHC II−ItgaxcreH2-Ab1c Fr D pre-B cells would contain cells that had not yet expressed MHC II, as well as more differentiated cells that were nevertheless MHC II− owing to H2-Ab1 deletion. These cells were transcriptionally indistinguishable from the less differentiated fraction of MHC II− WT Fr D pre-B cells (Figure 4A). MHC II–expressing ItgaxcreH2-Ab1c Fr D cells should represent a stage of differentiation equivalent to MHC II–expressing WT Fr D cells, also indicated by transcription of immunoglobulin genes (Figure 4A). However, expression of H2-Ab1 in ItgaxcreH2-Ab1c Fr D cells was much lower than in WT Fr D cells (Figure 4A), due to the presence of only 1 functional allele in the former cells (“Methods”). Notably, compared with WT counterparts, MHC II–expressing ItgaxcreH2-Ab1c Fr D cells exhibited reduced expression of other MHC II genes and lacked expression of the maturation markers that characterized the MHC II–expressing WT Fr D cells (Figure 4A; supplemental Figure 6B). These transcriptional differences were indicative of stunted or delayed differentiation and were also evident in the ItgaxcreH2-Ab1c Fr D population as a whole, which was transcriptionally distinct from the WT Fr D population (Figure 4C; supplemental Table 3).
Specific to ItgaxcreH2-Ab1c Fr D cells were a set of genes, including Cox6a2, Plcb4, and Erdr1 (Figure 4C), which were shared with common lymphoid progenitor (CLP) or Fr A cells (Figure 4D), suggesting retention of precursor traits in MHC II–deleted pre-B cells. Indeed, comparison of genes differentially expressed between ItgaxcreH2-Ab1c and WT Fr D B cells with precompiled expression profiles of developing B cells confirmed the advanced differentiated state of MHC II–expressing WT Fr D cells and, importantly, revealed that the respective ItgaxcreH2-Ab1c Fr D subset exhibited a transcriptional signature comparable with those of earlier precursors (CLP and Fr A) (Figure 4E; supplemental Figure 7). Additionally, whereas WT Fr D cells had become quiescent, as expected,1,34 ItgaxcreH2-Ab1c Fr D maintained a proliferative signature (Figure 4F). Thus, expression of MHC II at the earliest B-cell developmental stage appeared to promote B-cell differentiation at the expense of proliferation. These effects of MHC II appeared restricted to developing B cells, as ItgaxcreH2-Ab1c and WT splenic B cells were comparable both in terms of BCR repertoire diversity (supplemental Figure 8) and transcriptional signature, with the possible exception of Plcb4 and Erdr1 (Figure 4C). Lastly, the less differentiated transcriptional profile of MHC II–deficient Fr D pre-B cells was not due to misidentification of earlier stages, as the ratio of developing fractions was preserved in MHC II–deficient mice and cells phenotypically resembling Fr D pre-B cells were not present in Rag1−/−H2dlAb1-Ea mice, where B-cell differentiation is blocked at Fr C, owing to the lack of a functional BCR (supplemental Figure 9A).
Ectopic MHC II expression is sufficient to drive B-cell development
The early advantage of MHC class II–deleted B cells was still evident in competition between Rag1−/− B cells and Rag1−/−H2dlAb1-Ea B cells, which were again overrepresented in such mixed bone marrow chimeras (supplemental Figure 9B), despite a block in differentiation at Fr C. Similarly, transcription of Il7r, as well as known targets of interleukin-7 (IL-7) receptor signaling were broadly comparable between ItgaxcreH2-Ab1c and WT Fr D B-cell precursors (supplemental Figure 10). These data argued against an IL-7–driven mechanism or a requirement for BCR-initiated signals.
MHC II deficiency has been reported to enhance and reduce responsiveness of mature B cells and DCs to Toll-like receptor 4 (TLR4) agonists.15,16 Nevertheless, responsiveness to ligation of the 2 TLRs highly expressed in B-cell precursors, namely TLR2 and TLR4, was minimally affected by MHC II deficiency (supplemental Figure 11). These results raised the possibility that, instead of modifying responsiveness to known B-cell differentiation signals, MHC II was promoting precursor cell differentiation directly. To explore this possibility, we measured the efficiency of differentiation of HSCs ectopically expressing MHC II. Strikingly, in competitive in vitro B-cell differentiation assays, MHC II–transduced H2dlAb1-Ea HSCs exhibited considerably enhanced ability to differentiate, compared with H2dlAb1-Ea HSCs or WT HSCs, which did not yet endogenously express MHC II (Figure 5). Thus, ectopic MHC II expression was sufficient to promote B-cell differentiation of HSCs.

Ectopic MHC II expression drives B-cell development. (A) Competitive in vitro differentiation assay between CD45.1+ WT HSCs transduced with green fluorescent protein (GFP), CD45.2+H2dlAb1-Ea HSCs transduced with GFP, and CD45.2+H2dlAb1-Ea HSCs transduced with MHC II Ab1 and Aa chains. MHC II expression is shown 2 days after transduction of the respective HSCs, distinguished by GFP and CD45 allotype expression. (B) Expression of B-cell lineage markers B220, CD24, and PB-1 and percentage of lineage-negative (Lin−) and Fr A-C cells in each of the HSC donors described in panel A, following growth in B-cell media (n = 2). FCS-A, forward scatter area.
Ectopic MHC II expression drives B-cell development. (A) Competitive in vitro differentiation assay between CD45.1+ WT HSCs transduced with green fluorescent protein (GFP), CD45.2+H2dlAb1-Ea HSCs transduced with GFP, and CD45.2+H2dlAb1-Ea HSCs transduced with MHC II Ab1 and Aa chains. MHC II expression is shown 2 days after transduction of the respective HSCs, distinguished by GFP and CD45 allotype expression. (B) Expression of B-cell lineage markers B220, CD24, and PB-1 and percentage of lineage-negative (Lin−) and Fr A-C cells in each of the HSC donors described in panel A, following growth in B-cell media (n = 2). FCS-A, forward scatter area.
MHC II controls growth and differentiation of transformed B cells
The loss of MHC II is a frequent feature of B-cell malignancies and is associated with poor prognosis.5 MHC II–deleted B-cell precursors retain stem cell characteristics, favor self-renewal and overexpress cancer-associated genes such as phospholipase b4 (Plcb4)39 and erythroid differentiation regulator 1 (Erdr1).40,41 Moreover, many B-cell tumors rely on aberrant activation signals,42 which may be modified by MHC II. We therefore hypothesized that MHC II loss in transformed B cells may confer a cell-intrinsic fitness advantage, in addition to escape from CD4+ T-cell immune surveillance.
To explore this, we examined a series of precursor B-cell leukemia cell lines, originally developed in IL-7–overexpressing mice.43 Individual cell lines were arrested at distinct stages of B-cell development and exhibited variable MHC II expression (supplemental Figure 12A-B). Also variable was the expression of genes that characterized MHC II–deleted nontransformed pre-B cells, with the exception of Erdr1, which was highly expressed in all cell lines (supplemental Figure 12C). Sublines of A1, F6, or G7 leukemic cells selected by cell-sorting for high MHC II expression quickly became MHC II− upon in vivo growth in lymphocyte-deficient hosts (Figure 6A), signifying a possible growth advantage of MHC II− cells, independently of CD4+ T-cell immune surveillance.

MHC II controls growth and differentiation of transformed B cells. (A) Frequency of MHC II− leukemic cells recovered from Rag1−/− recipients that had received A1, F6, or G7 cells, sorted on the basis of MHC II expression prior to inoculation. (B) B3, D5, F6, or G7 cells, transduced with either GFP or MHC II Ab1 and Aa chains were mixed in equal ratios and transferred into Rag1−/− recipients. GFP and MHC II expression in mixtures of F6 cells before (pre) and after (post) transfer in Rag1−/− recipients is shown (left). Also shown is the frequency of GFP+MHC II− leukemic cells recovered from Rag1−/− recipients that had received mixtures of GFP or MHC II–transduced B3, D5, F6, or G7 cells (right). *P < .001 by Student t tests. (A-B) Symbols represent individual recipient mice and group means (plus or minus SEM) are also shown. (C) B3 and F6 cells were transduced either with GFP or MHC II Ab1 and Aa chains. GFP+ and MHC II+ cells from the same cell line were mixed at equal ratios and cocultured. Mean change (plus or minus SEM) from starting frequency of MHC II− cells over time in such cocultures (B3, n = 2; F6, n = 4). *P = .03; **P = .08 by 1-way ANOVA on ranks. (D) Growth rate, indicated by the specific growth rate constant, μ, of the exponential growth phase as measured with the IncuCyte live cell analysis system, of B3 or F6 cells transduced either with GFP or MHC II Ab1 and Aa chains. Symbols represent separate cultures. *P < .001 by 2-tailed Mann-Whitney rank-sum tests. (E) CD24 and BP-1 expression in parental F6 cells and those transduced either with GFP or MHC II Ab1 and Aa chains. (F) Expression of differentially expressed genes (at least twofold, q = 0.05) between MHC II− and MHC II+ F6 cells (left), expression of selected MHC II, immunoglobulin, and B-cell differentiation genes in the same cells (middle), and expression of selected genes downregulated in MHC II+ F6 cells (right).
MHC II controls growth and differentiation of transformed B cells. (A) Frequency of MHC II− leukemic cells recovered from Rag1−/− recipients that had received A1, F6, or G7 cells, sorted on the basis of MHC II expression prior to inoculation. (B) B3, D5, F6, or G7 cells, transduced with either GFP or MHC II Ab1 and Aa chains were mixed in equal ratios and transferred into Rag1−/− recipients. GFP and MHC II expression in mixtures of F6 cells before (pre) and after (post) transfer in Rag1−/− recipients is shown (left). Also shown is the frequency of GFP+MHC II− leukemic cells recovered from Rag1−/− recipients that had received mixtures of GFP or MHC II–transduced B3, D5, F6, or G7 cells (right). *P < .001 by Student t tests. (A-B) Symbols represent individual recipient mice and group means (plus or minus SEM) are also shown. (C) B3 and F6 cells were transduced either with GFP or MHC II Ab1 and Aa chains. GFP+ and MHC II+ cells from the same cell line were mixed at equal ratios and cocultured. Mean change (plus or minus SEM) from starting frequency of MHC II− cells over time in such cocultures (B3, n = 2; F6, n = 4). *P = .03; **P = .08 by 1-way ANOVA on ranks. (D) Growth rate, indicated by the specific growth rate constant, μ, of the exponential growth phase as measured with the IncuCyte live cell analysis system, of B3 or F6 cells transduced either with GFP or MHC II Ab1 and Aa chains. Symbols represent separate cultures. *P < .001 by 2-tailed Mann-Whitney rank-sum tests. (E) CD24 and BP-1 expression in parental F6 cells and those transduced either with GFP or MHC II Ab1 and Aa chains. (F) Expression of differentially expressed genes (at least twofold, q = 0.05) between MHC II− and MHC II+ F6 cells (left), expression of selected MHC II, immunoglobulin, and B-cell differentiation genes in the same cells (middle), and expression of selected genes downregulated in MHC II+ F6 cells (right).
To prevent the natural loss of MHC II expression in leukemic cells grown in vivo and to introduce MHC II expression in those cells that were previously negative, such as the B3 cells, we transduced them with H2-Ab1 and H2-Aa transgenes. Upon transplantation at equal ratios into lymphocyte-deficient hosts, MHC II− B3, F6, or G7 leukemic cells quickly outcompeted their MHC II–transduced counterparts, although in vivo growth of D5 leukemic cells remained unaffected (Figure 6B). Moreover, MHC II− F6 and B3 leukemic cells could outpace their MHC II–expressing equivalents in vitro, both in feeder cell-dependent and -independent growth conditions (Figure 6C-D).
Notably, transduction of F6 cells with MHC II also led to their phenotypic progression to Fr C (Figure 6E), further supporting a role for MHC II in promoting differentiation. Consistent with reduced in vitro and in vivo growth, MHC II–transduced F6 cells exhibited distinct changes in gene expression, including upregulated expression of additional MHC II–related genes, immunoglobulin genes, and other maturation markers (Figure 6F). Enhanced differentiation of MHC II–transduced F6 cells was associated with reduced expression of a smaller set of genes, including metabolic genes, such as Acot6, Galc, and P4htm (Figure 6F). These changes included modulation of fibroblast activation protein (Fap) transcription, encoding the dipeptidyl peptidase and endopeptidase FAP. Assessment of publicly available data38 revealed Fap expression in the earliest CD34− and CD34+ long-term murine HSCs (supplemental Figure 13). Moreover, immune targeting of FAP in mice uncovered expression in multipotent bone marrow stromal cells.44 Together, these results linked lack of MHC II expression in leukemic B-cell precursors with cell-intrinsic proliferative advantage and retention of a less differentiated phenotype.
Discussion
MHC II is of paramount importance in the initiation of adaptive immunity and is the strongest genetic determinant of susceptibility to autoimmune and infectious diseases.45-47 Although antigen presentation to CD4+ T cells is its primary function, accumulated evidence for signaling capacity of MHC II molecules suggests additional cell-intrinsic functions, which remain incompletely understood. The data presented here demonstrate that MHC II participates in the differentiation program of HSCs committed to the B-cell lineage, as well as in the maintenance of the differentiated phenotype of malignant B cells.
Despite a wealth of data demonstrating an effect of MHC II crosslinking on proliferation, differentiation, cellular adhesion, or cell death in a variety of in vitro model systems,7-11 a cell-intrinsic role for MHC II expression in hematopoietic cell survival or differentiation has remained elusive. With the obvious exception of MHC II–restricted CD4+ T cells and cell types that depend on CD4+ T cells for help, such as GC B cells, hematopoietic development of most cell types appears unaffected in MHC II–deficient mice.26,32 Although numbers of peripheral B cells are severely reduced in mice lacking expression of the MHC II α chain selectively,48 this effect is likely due to toxic accumulation of mispaired MHC II β chains.32 Such toxicity impaired survival of mature B cells, but did not appreciably affect B-cell development.32,48 Moreover, the limited analysis of mice bearing MHC II β chains with truncated intracellular domains failed to reveal differences in hematopoietic development.49 Similarly, our analysis of mice with germline of MHC II genes or conditional deletion of the H2-Ab1 gene showed normal representation of mature B-cell subpopulations (with the exception of GC B cells), as well as the BCR repertoire.
Not captured by these analyses, however, are effects of MHC II expression on cellular fitness. This can only be revealed in the context of competition between nonidentical cells with respect to MHC II expression. Heterogeneous MHC II expression would arise naturally when MHC II expression is developmentally initiated, such as, for example, in Fr D B cells. It also characterizes cancer evolution, with the emergence and eventual outgrowth of MHC II− leukemic cells. Indeed, our results with competitive bone marrow chimeras in vivo suggest that MHC II expression delivers a differentiation signal during the developmental stages at which MHC II is first expressed, further supported by direct in vitro evidence. Conversely, lack of MHC II expression promotes retention of self-renewal potential and confers a proliferative advantage in developing B cells.
Progressive upregulation of MHC II expression during B-cell development indicates a potential positive feedback loop consolidating B-cell maturation, with MHC II–derived differentiation cues progressively amplified. Notably, MHC II expression in pro-B cells appears developmentally regulated also at the organismal level and occurs during adult, but not fetal or neonatal, murine B-cell development.35,36 Also developmentally regulated is the balance of B1 and B2 B-cell differentiation, with a shift in favor of the latter in the adult.35,36 These observations support a model whereby MHC II− fetal or neonatal pro-B cells produce mature B1 B cells, whereas MHC II–expressing adult pro-B cells are progenitors of conventional B2 B cells.35,36
Depending on the cell type, MHC II crosslinking in vitro has been previously reported to trigger 2 major signaling cascades.8,10 One involves elevation of cellular cAMP levels and translocation of protein kinase C, and requires the MHC II β-chain cytoplasmic domain.8,10 Another leads to Src kinase activation and Ca2+ mobilization, entirely independently of the cytoplasmic domains of either MHC II chain.8,10 In vitro studies using mature B-cell lines have implicated an array of signal transducers or adaptors, including CD19, CD79, and STING, in MHC II–initiated signaling.7-11 However, most of these adaptors are not yet expressed in pro-B or pre-B cells, making them unlikely candidates, with the exception of STING (encoded by Tmem173), which is highly expressed at early stages of B-cell development. Moreover, our preliminary experiments with truncation of the cytoplasmic domains of either the MHC II Aα or Aβ chain failed to support a role for these domains in developing B cells and it is currently unclear whether the effects of MHC II expression described in this study involve known MHC II–signaling cascades.
MHC II deficiency in nontransformed B-cell precursors is associated with transcriptional induction of genes, such as Plcb4 and Erdr1, that are linked to retention of stem cell properties or cellular transformation, but are not normally expressed in healthy B cells. Plcb4 is involved in cellular senescence, and gain-of-function mutations in Plcb4 are a primary cause of uveal melanoma in humans.39 The mouse-specific Erdr1 gene was first discovered in murine B-cell leukemia lines, where it functions as a stress-related survival factor,40,41 and we found that is also highly expressed in nontransformed CD34+ HSCs and MHC II–deficient bone marrow B-cell progenitors. The expression pattern of Erdr1 has remained largely underreported, partly due to its incomplete genomic annotation. Our expression database search indicated that the expression of Erdr1 (annotated as the Y-linked Gm47283 gene) mirrors expression of Gm21887 (an X-linked gene). Based on gene location, structure, and sequence inspection, we conclude that Gm21887 is part of an X-Y syntenic region and should therefore be considered an Erdr1 paralogue. In contrast to the mouse genome that contains 2 copies of Erdr1, and despite earlier reports suggesting the existence of a human ERDR1 gene coding a protein identical to the murine equivalent,40 no ortholog of the murine Erdr1 gene is present in the current assembly of the human genome (GRCh38.p12). As cell-autonomous effects of MHC II ligation have been described, at least in vitro, both in human and murine cells,8 the presence of Erdr1 in mice but not in humans, makes it an unlikely candidate to mediate these effects.
Despite the upregulation of cancer-associated genes in MHC II–deficient, but otherwise normal, B-cell precursors, MHC II deficiency did not lead to tumor development in the strains and timeframe we have analyzed. Nevertheless, our data revealed a clear effect of MHC II expression in restricting precursor B-cell leukemia growth, both in vitro and in vivo, independent of CD4+ T-cell immunosurveillance. Although induced in a variety of hematopoietic and nonhematopoietic cells during inflammation, MHC II expression is often lost following transformation, particularly in certain B-cell malignancies.5,50-52 Traditionally considered an escape mechanism from CD4+ T-cell immunosurveillance, our findings suggest that loss of MHC II expression in transformed cells may additionally provide a cell-intrinsic fitness advantage by virtue of reduced differentiation. Indeed, lack of MHC II expression was associated with retention of stem cell characteristics, indicating that MHC II deficiency may have far wider effects on cellular dedifferentiation. Selection of cells retaining stem cell properties, as well as escaping immunosurveillance following loss of MHC II would undoubtedly accelerate cancer progression.
The RNAseq data reported in this article have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress; accession numbers E-MTAB-6763 and E-MTAB-6765).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Amanda Fisher for providing the murine bone marrow B-cell leukemia cell lines, and Jakob Loschko and Michel C. Nussenzweig for providing the Zbtb46creH2-Ab1c mice. The authors are grateful for assistance from the Flow Cytometry, Biological Resource, Cell Services, and High Throughput Screening facilities at the Francis Crick Institute. This work benefited from data assembled by the ImmGen and Cancer Cell Line Encyclopedia (CCLE) consortia.
This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001099), the UK Medical Research Council (FC001099), and the Wellcome Trust (FC001099).
Authorship
Contribution: J.M. designed, performed, and analyzed most of the experiments with assistance from U.E. and L.D.; J.A. and G.R.Y. assisted in the acquisition and analysis of RNAseq data; C.N. and P.T. performed the large-scale fluorescence imaging; and G.K. conceived and supervised the project and, together with J.M., wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: George Kassiotis, The Francis Crick Institute, 1 Midland Rd, London NW1 1AT, United Kingdom; e-mail: george.kassiotis@crick.ac.uk.


