Key Points
ADAMTS13 structure is exceptionally variable among vertebrates.
Functional, fully allosteric regulated ADAMTS13 requires no more than 3 distal thrombospondin repeats.

Abstract
The metalloprotease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats member 13) prevents microvascular thrombosis by cleaving von Willebrand factor (VWF) within platelet-rich thrombi, and cleavage depends on allosteric activation of ADAMTS13 by the substrate VWF. Human ADAMTS13 has a short propeptide, metalloprotease (M), disintegrin-like (D), thrombospondin-1 (T), Cys-rich (C), and spacer (S) domains (proximal domains), followed by 7 T and 2 CUB (complement components C1r and C1s, sea urchin protein Uegf, and bone morphogenetic protein-1) domains (distal domains). Distal domains inhibit the catalytic proximal domains; binding of distal T8-CUB domains to the VWF D4 domain relieves autoinhibition and promotes cleavage of the nearby VWF A2 domain. However, the role of specific ADAMTS13 distal domains in this allosteric mechanism is not established. Assays of plasma ADAMTS13 from 20 placental mammals, birds, and amphibians show that allosteric regulation is broadly conserved, and phylogenetic analysis of 264 vertebrates shows the long propeptide, T3, T4, T6, and T6a domains have been deleted several times in placental mammals, birds, and fish. Notably, pigeon ADAMTS13 has only 3 distal T domains but was activated normally by human VWF D4 and cleaved VWF multimers, preferentially under fluid shear stress. Human ADAMTS13 constructed to resemble pigeon ADAMTS13 retained normal allosteric regulation and shear-dependent cleavage of VWF. Thus, the T3-T6 domains of human ADAMTS13 are dispensable. Conversely, deletion of T7 or T8 abolished allosteric activation. For most species, some sequence changes in the VWF substrate can markedly increase the rate of cleavage, suggesting that ADAMTS13 and VWF have not evolved to be optimal enzyme-substrate pairs. These properties may reflect evolutionary pressure to balance the risk for VWF-dependent bleeding and thrombosis.
Introduction
von Willebrand factor (VWF) is a multimeric protein that tethers platelets to endothelial lesions and initiates the repair of damaged vascular tissue.1 During hemostasis, the metalloprotease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats member 13) prevents excessive platelet adhesion by cutting a cryptic site within the VWF A2 domain and dissolving VWF-platelet aggregates.2 Genetic or autoimmune deficiency of ADAMTS13 impairs this regulatory mechanism and causes thrombotic thrombocytopenic purpura (TTP), which is characterized by life-threatening microvascular thrombosis.3
Human ADAMTS13 is a multidomain protein with a propeptide, metalloprotease (M), disintegrin-like (D), thrombospondin-1 (T), Cys-rich (C), and spacer (S) domains, followed by 7 T and 2 CUB (complement components C1r and C1s, sea urchin protein Uegf, and bone morphogenetic protein-1) domains.4,5 Unlike other ADAMTS family members,6 human ADAMTS13 has a relatively short propeptide of 41 amino acid residues; it is roughly 20% the size of an average ADAMTS propeptide, and plays no role as an intramolecular chaperone or in the maintenance of latency for the newly synthesized protease.7 In fact, human ADAMTS13 with or without a propeptide is secreted efficiently and has enzymatic activity toward VWF.8 The metalloprotease domain possesses an active site motif (HEXXHXXGXXHD) that coordinates a Zn2+ ion.5,9 The disintegrin-like domain has a hydrophobic pocket that may be essential for ADAMTS13 interaction with the VWF A2 substrate.10 Optimal cleavage of VWF by ADAMTS13 also requires the spacer domain, which contains an exosite that directly binds to the C-terminal end of unfolded VWF A2.11,12 This exosite is the most common target of inhibitory autoantibodies (auto-Abs) in patients with TTP.12-15
The functions of the distal domains (T2-CUB) are less well characterized. Deletion of distal domains results in an ADAMTS13 that is hyperactive toward short peptide substrates, but impairs its ability to bind and cleave multimeric VWF under flow.16-19 These opposite effects are explained in part by the binding of ADAMTS13 distal domains, particularly T8, to VWF domain D4. This binding interaction may position ADAMTS13 favorably to recognize and cleave the adjacent VWF domain A2.20
We21 and others22 recently uncovered another allosteric function of ADAMTS13 distal domains. We showed that ADAMTS13 distal domains fold back to interact with proximal domains and inhibit proteolytic activity. This autoinhibition is relieved by some monoclonal Abs (mAbs) that bind to specific distal domains, by binding to VWF D4, by lowering the pH to pH 6, or by deletion of specific distal domains. South and colleagues arrived at similar conclusions after demonstrating that isolated recombinant distal ADAMTS13 domains inhibited a hyperactive variant of ADAMTS13 that lacked distal domains.22
We now show that ADAMTS13 allostery is broadly conserved among vertebrates, with a few potential exceptions. The distal T3-T6 domains are dispensable for normal allosteric regulation, whereas T7, T8, and CUB domains are required. Even though ADAMTS13 from different species exhibits striking variation in substrate specificity, human recombinant VWF D4 can allosterically activate almost all of them. Furthermore, susceptibility to allosteric activation confers increased specificity for the cleavage of multimeric human VWF under shear stress.
Methods
Sequences and Analysis
ADAMTS13 genomic DNA and cDNA sequences were assembled for 206 species by BLASTN and TBLASTN searching of GenBank assemblies, trace archives, and Sequence Read Archive transcriptomic and genomic data, using the Lasergene programs EditSeq, MegAlign, SeqBuilder, and SeqMan Pro (Version 14.0.0, DNASTAR, Madison, WI). Exons were identified by the alignment of transcriptomic and genomic sequences. Conserved domains were identified by reverse position-specific BLAST, as described previously.23 Translated protein sequences were aligned using the Clustal W method, Gonnet protein weight matrices, gap penalty 10, and gap length penalty 0.2.
Blood plasma samples
Heparinized pooled human plasma (>35 donors) was purchased (Biological Specialty Corp., Colmar, PA). Banked human plasma samples were obtained under the protocol approved for the ART study (Adjuvant Low Dose Rituximab for Acquired TTP with Severe ADAMTS13 Deficiency; NCT01554514). Blood and tissue specimens from animals were discarded materials donated after routine veterinary evaluations or were purchased from BiolVT.
Recombinant ADAMTS13
Wild-type ADAMTS13 and ADAMTS13 truncated after domain S (MDTCS), T7 (MT7), or T8 (MT8) have been described.21 ADAMTS13 with internal deletion of T2-T7 (Trp686-Arg1075; delT2-7) and T3-T6 (Trp746-Arg1015, delT3-6) were engineered by mutagenesis of wild-type ADAMTS13 in pTriEx-7 Ek/LIC (EMD Millipore). Constructs encode a mouse IgM signal peptide, StrepTag II, and enteropeptidase recognition sequence followed by Ala75 of ADAMTS13. Pigeon, horse, and armadillo ADAMTS13 constructs were synthesized (Genewiz, South Plainfield, NJ) and cloned in pTriEx-7 Ek/LIC. Two pigeon ADAMTS13 variants were constructed that contain the pTriEx-7 signal peptide, N-terminal StrepTag II and enteropeptidase epitope tags; 1 (proADAMTS13) follows with the initiation codon and the other (mature ADAMTS13) follows with the first residue after the furin cleavage site (Ala201). Proteins were stably expressed in T-REx 293 cells (Invitrogen) grown for 48 hours in Freestyle 293 serum-free media (Gibco), and chromatographically purified as described.21 Some 293 cells expressing pigeon ADAMTS13 were grown in the presence of 50 µM decanoyl-Arg-Val-Lys-Arg-chloromethane (Cayman Chemical, Ann Arbor, MI). Protein concentration was determined by the BCA method (Pierce, Thermo Fisher).
ADAMTS13 fluorogenic peptide substrates
FRETS-rVWF71 (human VWF71) was prepared as described.24 Rat, cattle, armadillo, and pigeon variant VWF71 substrates were prepared. The final constructs in plasmid pET32Xa encode thioredoxin, a His-tag, a TEV cleavage site, a Gly residue, and species-specific VWF residues corresponding to human VWF residues Gln1599-Arg1668. Cattle and rat protein expression, purification, thioredoxin cleavage, peptide purification, and labeling were performed as described for human VWF71.24 Purified armadillo or pigeon peptides were labeled with the 2 dyes simultaneously in 50 mM sodium phosphate at pH 7.4, for 1 hour, followed by high-performance liquid chromatography purification of the doubly labeled product. Rat and pigeon VWF71 unlabeled peptides were poorly soluble and yielded enough substrate for relatively few assays.
ADAMTS13 mAbs and VWF D4
ADAMTS13 assays with fluorogenic substrates
The concentration of fluorogenic substrates was determined by absorbance at 629 and 819 nm.24 Standard plasma ADAMTS13 assays21 included 50 mM Bis-Tris (pH 6) or HEPES (pH 7.4), 150 mM NaCl, 10 mM CaCl2, 0.05% Tween-20, 1 mg/mL bovine serum albumin, 1 µM fluorogenic substrate, 100 µM phenylmethylsulfonyl fluoride, 3 µL HALT protease inhibitor cocktail (Thermo Fisher Scientific), and plasma or recombinant ADAMTS13 variants in a total volume of 200 µL. Negative control assays were performed similarly with added 10 mM EDTA or indicated amounts of mAb 3H9. Reaction was initiated by adding substrate to other assay components in 96-well white microplates (Optiplate-96, PerkinElmer) at 30°C. Increasing fluorescence was monitored with a Victor2V Multilabel Counter (PerkinElmer) or Synergy H1 Hybrid Multi-Mode Microplate Reader (Biotek) with 635 ± 10-nm excitation and 660 ± 10-nm emission filters. Assays by this method have an interassay coefficient of variation of less than 2%.24
ADAMTS13 in animal plasmas was screened for activation by anti-human ADAMTS13 mAbs 7C4, 19H4, and 12D4 (∼2 µg/reaction) or VWF D4 (up to 20 µM) in assays with human VWF71. Differences between mean values were assessed with the unpaired Student t test.
Cleavage of VWF Multimers
Cleavage of VWF by ADAMTS13 and variants was performed as described, with modifications.21 In brief, using 1.5-mL microcentrifuge tubes and 60-µL total reaction volume, 40 nM purified human plasma VWF (Haematologic Technologies, Essex Junction, VT) in 50 mM HEPES at pH 7.4, 150 mM NaCl, 5 mM CaCl2, and 1 mg/mL bovine serum albumin was vortexed with 50 nM ADAMTS13 variants, with or without 10 mM EDTA. It should be noted that enzyme turnover should not be achieved under these conditions, a limitation of this assay. Samples (1-10 µL) were mixed with Novex 2X Tris-glycine or NUPAGE 4X LDS sample buffer, heated at 70°C for 10 minutes, and then electrophoresed on a Novex 4% Tris-glycine gel, 150 V for 90 minutes; 3% to 8% Tris-Acetate gel, 150 V for 60 minutes; or Novex WedgeWell 6% Tris-Glycine gel, 225 V for 40 minutes. Gels were incubated in equilibration buffer (2× NuPAGE transfer buffer containing 10% methanol and 1:1000 NuPAGE antioxidant; Thermo Fisher Scientific) for 20 minutes, and rinsed 3 times in water, and finally in equilibration buffer without antioxidant. Proteins were transferred onto a polyvinylidene fluoride membrane for 10 minutes at 23 V, using an iBlot device (Life Technologies), or for 90 minutes at 25 V in a wet transfer mini gel tank (Invitrogen). The membrane was air dried for 30 to 60 minutes, reactivated with 100% methanol, washed 3 times for 5 minutes in water, blocked for 2 hours with Odyssey blocking TBS (Tris-buffered saline) buffer (LICOR, Lincoln, NE), and incubated overnight in casein blocking buffer containing 1:10 000 rabbit anti-VWF Ab 082 (Dako). The membrane was rinsed 4 times with TBST (TBS with tween-20) and incubated with secondary Ab IRDye 680RD (fluorescent dye labeled goat anti-rabbit, 1:15 000) in TBST supplemented with 0.007% sodium dodecyl sulfate for 2 hours. The membrane was washed 4 times with TBST. Protein bands were imaged with an Odyssey CLx and analyzed with Image Studio 5.x (Li-COR).
Binding Assays
Dissociation constants (Kd) for ADAMTS13 variants binding to recombinant VWF D4-CK domains were determined by biolayer interferometry, using an Octet RED96 (ForteBio) as described.21
Results
Some ADAMTS13 domains have been lost or modified during evolution
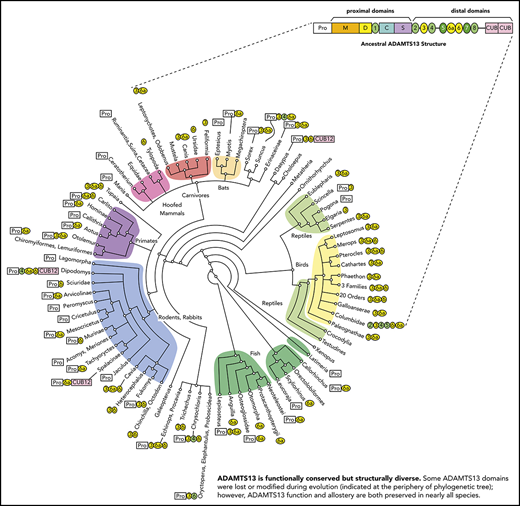
We characterized the variations in ADAMTS13 domain structure, using phylogenetic analysis. GenBank contains experimentally determined ADAMTS13 cDNA sequences for only human, mouse, and dog. Computationally assembled ADAMTS13 cDNA sequences were available for another ∼200 vertebrates, but only 56 of them were free of major errors. Therefore, we assembled and manually curated complete ADAMTS13 cDNA and genomic DNA sequences from publicly available transcriptomic and genomic data. Two hundred sixty-four species (supplemental Table 1, available on the Blood Web site) were selected to represent monotremes, placental and marsupial mammals, cartilaginous and bony fish, birds, amphibians, and several kinds of reptile.
Sequence comparisons (Figure 1; supplemental Figure 1; supplemental Table 1) show that the human ADAMTS13 domain structure is fully shared by less than 25% of the species studied, with several domains having been lost or modified repeatedly during evolution. Ancestral ADAMTS13 was defined as the sequence containing the largest domain structure that is found today in early progenitors (eg, amphibians), but still preserved in some mammals (eg, horse). The ancestral ADAMTS13 had an extra distal T domain (T6a) and a long propeptide of ∼120 to ∼200 amino acid residues that is characteristic of most metalloproteases, but is notably missing from human ADAMTS13. Today, the ancestral structure is found in crocodilians, turtles, amphibians, marsupials, monotremes, horse, ferret, 2-toed sloths (Choloepus), and flying lemurs (Galeopterus).
Evolutionary variation in ADAMTS13 structure. The domain architecture of ADAMTS13 was determined for 264 species listed in supplemental Table 1. The apparent ancestral structure, with a long propeptide and a total of 9 T domains, is indicated at the top. Domains T3, T6a, and T6 (shaded yellow) have been deleted independently several times in different lineages. Other representative structures are shown for selected vertebrates. Names in bold indicate species or groups from which plasma samples were available for ADAMTS13 assays.
Evolutionary variation in ADAMTS13 structure. The domain architecture of ADAMTS13 was determined for 264 species listed in supplemental Table 1. The apparent ancestral structure, with a long propeptide and a total of 9 T domains, is indicated at the top. Domains T3, T6a, and T6 (shaded yellow) have been deleted independently several times in different lineages. Other representative structures are shown for selected vertebrates. Names in bold indicate species or groups from which plasma samples were available for ADAMTS13 assays.
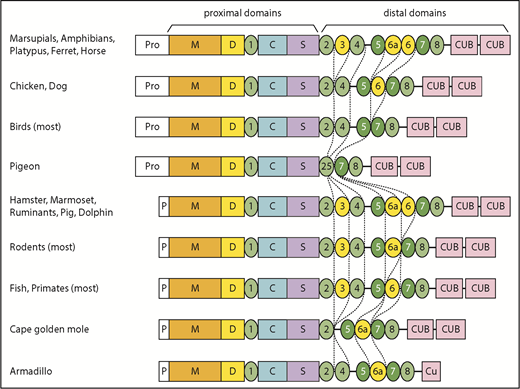
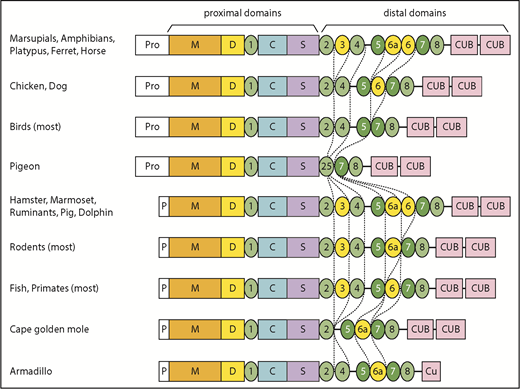
Some species in almost every lineage have independently deleted or inactivated exons 2a and 2b. These species include most fish and reptiles, as well as placental mammals except for horses, camelids (Tylopoda), carnivores, and fruit bats (Megachiroptera). Species without exons 2a and 2b have a short ADAMTS13 propeptide of ∼40 amino acids similar to that of human ADAMTS13. Whether short or long, the propeptide almost always has a potential furin cleavage site (supplemental Table 1).
In addition, T3, T6, and T6a have been deleted independently several times in placental mammals, birds, fish, and some reptiles. These particular T domains may be susceptible to loss because the exons that encode them have lengths that are multiples of 3 nucleotides and can be deleted without changing the reading frame.
More dramatic changes occur in a few species, such as the common pigeon (Columba livia). Similar to many birds, pigeon has lost T3, T6a, and T6. In addition, pigeon has lost exons 18 to 21, thereby deleting T4 and fusing the N-terminal part of T2 with the C-terminal part of T5 to make a complete T2/T5 hybrid domain (Figure 1; supplemental Figure 2B). As a result, pigeon ADAMTS13 has only 3 distal T domains (Figure 1). The extinct passenger pigeon (Ectopistes migratorius) had ADAMTS13 with the same structure (supplemental Table 1). A few placental mammals (eg, Cape golden mole) also have lost T4. Armadillo (supplemental Figure 2C) and Nannospalax galili ADAMTS13 are truncated after the first disulfide loop of CUB1, leaving one-half of a CUB domain, and Dipodomys ordii has no CUB domains at all (Figure 1; supplemental Table 1). Conversely, the proximal MDTCS and distal T7 and T8 domains are invariably present.
ADAMTS13 allostery is conserved in vertebrates
We investigated the enzymatic properties of plasma ADAMTS13 from 19 nonhuman species: dog, ferret, polar bear, armadillo, rabbit, primates (baboon, chimpanzee, marmoset, macaque), rodents (rat, mouse), ruminants (cattle, goat, sheep), pig, horse, birds (chicken, pigeon), and Xenopus laevis (Table 1). With 4 exceptions, these animal plasmas cleaved human VWF71 more slowly than did human plasma (Table 1). Interestingly, polar bear, armadillo, rabbit, and dog plasma were 2-fold to 4-fold more active than human plasma (Table 1). For every species, activity was dependent on divalent metal ions and resistant to inhibition by phenylmethylsulfonyl fluoride, as expected for orthologs of ADAMTS13. In addition, mAb 3H9, which recognizes the M domain of human ADAMTS13, prevented the cleavage of human VWF71 by plasma from primates (human, marmoset, macaque, baboon, and chimpanzee) and partially inhibited cleavage by plasma from dog, ferret, horse, pig, or polar bear (supplemental Table 2).
To characterize the allosteric properties of ADAMTS13 from these vertebrates, we determined whether cleavage of human VWF71 was increased by activators of human ADAMTS13: low pH, mAbs against T6-CUB domains, and VWF D4 (Table 1). Plasma ADAMTS13s from 12 species were activated ∼1.6-fold to ∼16-fold at pH 6.0 compared with pH 7.4. C57BL/6 mouse (known to carry an ADAMTS13 truncation after T6), ferret, and rabbit plasmas were relatively insensitive to low pH. Goat, chicken, dog, and armadillo were exceptional because they were markedly inhibited by low pH.
Except for C57BL/6 mouse, armadillo, and dog, all plasmas were activated by either mAbs or VWF D4 (Table 1). The most remarkable effect was observed for horse ADAMTS13, which was activated ∼26-fold by mAbs. Activating mAbs had no effect on ADAMTS13 for rat, chicken, pigeon, frog, ferret, polar bear, armadillo, rabbit, or dog, possibly because the mAbs do not recognize ADAMTS13 from these species. For example, they do not recognize recombinant armadillo or pigeon ADAMTS13 on western blots (data not shown).
Although the magnitude of activation varied, human VWF D4 increased the activity of ADAMTS13 from all species tested except mouse, armadillo, and dog. Failure to activate C56BL/6 mouse and armadillo ADAMTS13 is consistent with the lack of CUB domains in these species. In contrast, absence of all but 3 distal T domains in pigeon ADAMTS13 did not impair activation by D4. These results indicate that allosteric regulation of ADAMTS13 is highly conserved.
ADAMTS13 and VWF are not optimal enzyme-substrate pairs
To assess species-dependent preferences for VWF, we prepared fluorogenic substrates containing VWF sequences of cattle, rat, armadillo, and pigeon (Figure 2A). Compared with human VWF71, amino acid differences occur at 9 residues in armadillo and rat VWF71, at 13 residues in cattle VWF71, and at 34 residues in pigeon VWF71. Human, cattle, rat, and pigeon VWF-71 substrates have Tyr-Met cleavage sites, but armadillo VWF71 has a Tyr-Leu cleavage site.
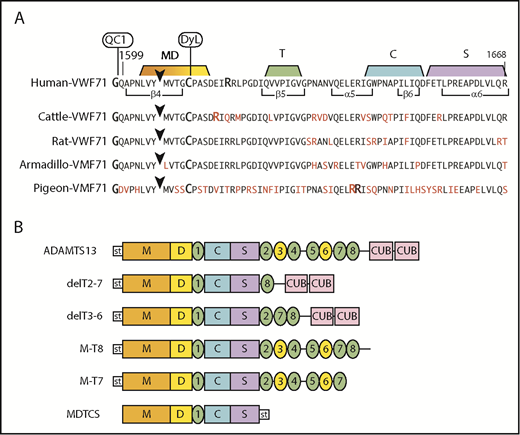
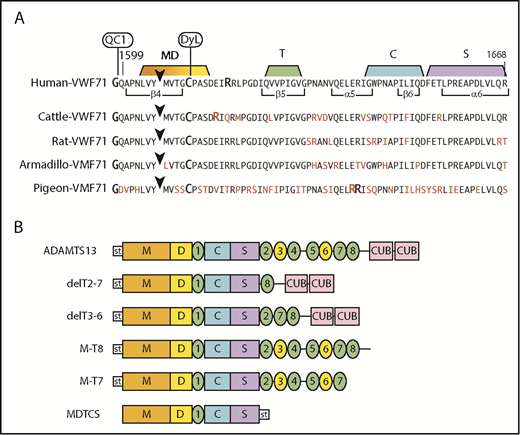
Fluorogenic substrates and ADAMTS13 constructs. (A) Substrates prepared from species-specific VWF sequences. The arrowhead marks the Tyr-Met bond (Armadillo, Tyr-Leu) cleaved by ADAMTS13. The N-terminus is modified with IRDye QC-1 (QC1), and the indicated Cys residue is modified with DyLight 633 (DyL). Segments of the sequence that make extensive contacts with MDTCS domains are indicated. Residues in red indicate differences from the human sequence. (B) Recombinant human ADAMTS13 and variants. Except for MDTCS, all constructs have an N-terminal StrepTag II (st) and enteropeptidase cleavage site.
Fluorogenic substrates and ADAMTS13 constructs. (A) Substrates prepared from species-specific VWF sequences. The arrowhead marks the Tyr-Met bond (Armadillo, Tyr-Leu) cleaved by ADAMTS13. The N-terminus is modified with IRDye QC-1 (QC1), and the indicated Cys residue is modified with DyLight 633 (DyL). Segments of the sequence that make extensive contacts with MDTCS domains are indicated. Residues in red indicate differences from the human sequence. (B) Recombinant human ADAMTS13 and variants. Except for MDTCS, all constructs have an N-terminal StrepTag II (st) and enteropeptidase cleavage site.
Armadillo and chicken plasma cleaved armadillo VWF71 and human VWF71 at approximately the same rate, suggesting little preference for a Tyr-Leu or Tyr-Met bond. Other species cleaved armadillo VWF71 more slowly, from 0% to 78% as rapidly as human VWF71. Pigeon ADAMTS13 cleaved pigeon VWF71 2.5-fold faster than human VWF71, and human ADAMTS13 was inactive toward pigeon VWF71 (data not shown). Only horse and FVB mouse plasma cleaved rat VWF71 faster (∼1.5-fold) than human VWF71. Interestingly, all species preferred cattle VWF71 to human VWF71, cleaving it from ∼1.5- to 15-fold faster (Table 2). Thus, ADAMTS13 exhibits substantial variation in recognition of VWF from other vertebrates, and some changes in the VWF sequence can markedly increase the rate of cleavage. This conclusion assumes that ADAMTS13 in various animal plasmas is at a similar level as in human plasma, and no alternative splicing or proteolytic variants are present.
Comparisons of recombinant and plasma ADAMTS13
Recombinant ADAMTS13 for armadillo, horse, human, and pigeon was expressed and purified (Figure 3). The proteins consist of a single main component of the expected size except for armadillo, which is a mixture of 2 major bands that share an N-terminal StrepTag II by immunoblotting (not shown). The upper band has the apparent mass (140 kDa) expected of full-length armadillo ADAMTS13. The lower ∼103-kDa band is consistent with proteolytic cleavage in the short linker between T4 and T5 (Figure 1). For each species, recombinant and plasma ADAMTS13 had similar relative activity toward human, cattle, and armadillo VWF71 substrates (Table 2).
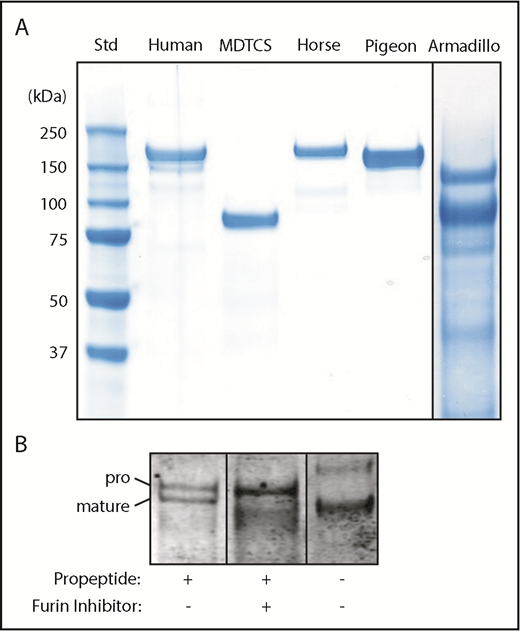
Recombinant ADAMTS13. (A) The indicated ADAMTS13 variants were expressed in 293T cells, purified and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and staining with InstantBlue (Expedion, San Diego, CA). Size in kilodaltons is indicated for standards. (B) Pigeon ADAMTS13 constructs with or without the propeptide were expressed in 293T cells in the presence or absence of the furin inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethane (50 µM), as indicated. Samples of conditioned medium were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting with anti-6xHis Ab. The positions of proADAMTS13 and mature ADAMTS13 are indicated.
Recombinant ADAMTS13. (A) The indicated ADAMTS13 variants were expressed in 293T cells, purified and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and staining with InstantBlue (Expedion, San Diego, CA). Size in kilodaltons is indicated for standards. (B) Pigeon ADAMTS13 constructs with or without the propeptide were expressed in 293T cells in the presence or absence of the furin inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethane (50 µM), as indicated. Samples of conditioned medium were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting with anti-6xHis Ab. The positions of proADAMTS13 and mature ADAMTS13 are indicated.
Full-length pigeon ADAMTS13 was secreted as 2 major species that differ by the presence or absence of the propeptide (Figure 3B). Inhibition of furin blocks propeptide cleavage resulting in secretion of predominantly proADAMTS13, and expression without the propeptide led to secretion of only mature ADAMTS13. All these proteins cleaved VWF71 at comparable rates and were activated ∼5-fold by VWF D4 (data not shown). Thus, as observed for the short 41-residue propeptide of human ADAMTS13,8 the long ∼176-residue propeptide of pigeon ADAMTS13 is not required for intracellular processing, secretion, proteolytic activity, or allosteric regulation.
Several distal TSP1 domains are dispensable for allosteric regulation of ADAMTS13
Analysis of 264 vertebrate ADAMTS13 sequences (supplemental Figure 1; supplemental Table 1) shows that T7 and T8 are universally conserved. CUB2 is present in all but 3 species; 1 of them also lacks CUB1, and the other 2 have a partial CUB1 domain. These conserved domains are likely to be essential for allosteric regulation. Conversely, many species have lost 1, several, or all of domains T3 through T6b, suggesting they are dispensable. These predictions are supported by mutagenesis of human ADAMTS13.
With respect to binding, human construct MT8 (Figure 2) bound to VWF D4-CK with a Kd of ∼620 nM, which is slightly higher than the Kd of ∼360 nM for ADAMTS13,21 whereas MT7 and delT2-7 did not bind to VWF D4-DK (Figure 4). Although delT2-7 possesses T8, lack of VWF binding may suggest that adjacent T domains induce conformational changes to facilitate binding. Thus, T7 and T8 appear necessary and CUB domains make a smaller contribution to binding VWF D4.
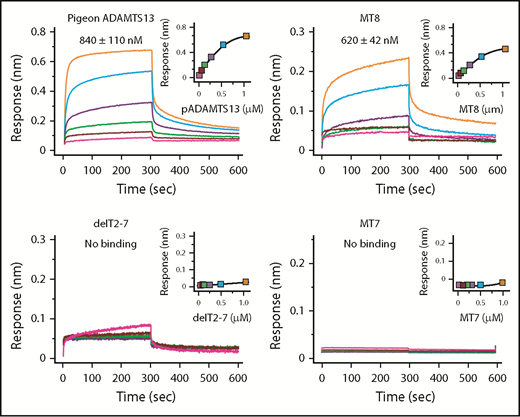
Binding of ADAMTS13 variants to VWF D4. Biolayer interferometry sensorgrams, steady-state analyses, and Kd values ± standard error are shown for the indicated ADAMTS13 proteins binding to His-tagged VWF D4-CK immobilized on Ni-NTA biosensors.
Binding of ADAMTS13 variants to VWF D4. Biolayer interferometry sensorgrams, steady-state analyses, and Kd values ± standard error are shown for the indicated ADAMTS13 proteins binding to His-tagged VWF D4-CK immobilized on Ni-NTA biosensors.
The same ADAMTS13 domains contribute to both autoinhibition and allosteric activation by VWF D4. For example, MT8 was partially autoinhibited, cleaving human VWF71 at a rate (∼4.0 min–1) between that of MDTCS (∼7.4 min–1) and ADAMTS13 (1.9 min–1). MT8 and ADAMTS13 were activated by VWF D4 ∼1.7-fold and ∼1.8-fold, respectively (Table 3), indicating that ADAMTS13 autoinhibition and allostery depend on T8 and at least 1 CUB domain, which has been identified as CUB1.27
Pigeon ADAMTS13 has only 1 distal T domain in addition to the invariant T7 and T8 (Figure 1), but has allosteric properties similar to human ADAMTS13. For example, recombinant pigeon ADAMTS13 bound to human VWF D4 with a Kd of ∼840 nM (Figure 4) and was activated ∼5-fold (Table 3) by human VWF D4, even more efficiently than full-length human ADAMTS13 (1.8-fold). These results suggest that several T domains of human ADAMTS13 are unnecessary, and this conclusion is supported by the effects of internal T deletions (Figure 2B). Construct delT2-7 did not bind VWF D4-CK (Figure 4) and was not activated by VWF D4 (Table 3). Restoring domains T2 and T7 in construct delT3-6 restored activation by VWF D4, suggesting that T7 and possibly T2 participate in binding to VWF, and that domains T3-T6 are dispensable.
Allosteric activation of ADAMTS13 facilitates VWF multimer cleavage
To assess allosteric regulation under more physiological conditions, we characterized the cleavage of multimeric VWF under shear stress by pigeon ADAMTS13 and selected variants of human ADAMTS13. Samples of plasma VWF and ADAMTS13 proteins were subjected to shear stress by vortexing, and cleavage products were characterized by western blotting and densitometry. The activity of each ADAMTS13 variant was normalized to concurrent assays with full-length recombinant human ADAMTS13 (Figure 5).
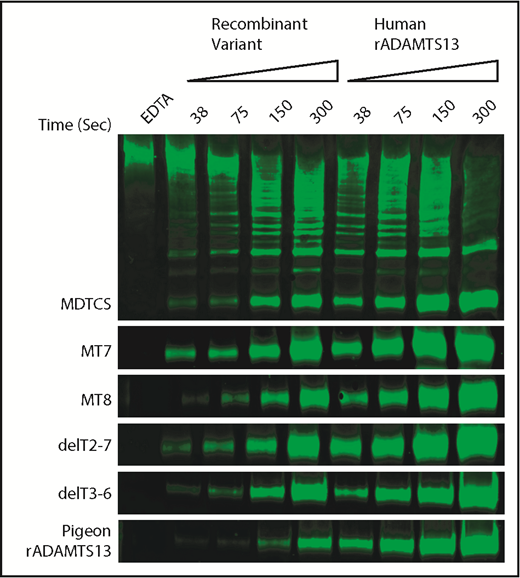
Shear-induced cleavage of VWF multimers by ADAMTS13. Human plasma VWF (40 nM) was sheared by vortexing for the indicated times with ADAMTS13 variants or human recombinant ADAMTS13 (rADAMTS13, 50 nM). Reactions were stopped by adding 10 mM EDTA. Cleavage products were analyzed by gel electrophoresis and western blotting with rabbit anti-VWF and goat anti-rabbit IRDye 680RD. The major 350-kDa dimeric VWF cleavage product was quantitated by fluorescence imaging. Cleavage rates for ADAMTS13 mutants were normalized to the cleavage rate for concurrent controls with human rADAMTS13. Data from duplicates are summarized in Table 3.
Shear-induced cleavage of VWF multimers by ADAMTS13. Human plasma VWF (40 nM) was sheared by vortexing for the indicated times with ADAMTS13 variants or human recombinant ADAMTS13 (rADAMTS13, 50 nM). Reactions were stopped by adding 10 mM EDTA. Cleavage products were analyzed by gel electrophoresis and western blotting with rabbit anti-VWF and goat anti-rabbit IRDye 680RD. The major 350-kDa dimeric VWF cleavage product was quantitated by fluorescence imaging. Cleavage rates for ADAMTS13 mutants were normalized to the cleavage rate for concurrent controls with human rADAMTS13. Data from duplicates are summarized in Table 3.
The results show a strong relationship between allosteric activation and preferential cleavage of VWF multimers under shear stress (Table 3). For example, VWF D4 does not activate MDTCS, MT7, or delT2-7, and these variants cleaved VWF multimers at ∼25% the rate of full-length ADAMTS13. In contrast, VWF D4 activates MT8 and delT3-T6, which cleaved VWF multimers ∼2-fold more rapidly than MDTCS, MT7, or delT2-7. These differences are more pronounced if the rate of multimer cleavage is normalized to the rate of VWF71 cleavage (Table 3). By this selectivity index (SI), allosterically regulated human ADAMTS13 (SI ∼54) is at least 10-fold more selective for VWF multimers than MDTCS, MT7, or delT2-7, which exhibit no allosteric regulation (SI ∼2 to ∼5).
Pigeon ADAMTS13 fits this pattern (Table 3). Recombinant pigeon ADAMTS13 cleaved human VWF71 ∼7% as rapidly as human ADAMTS13, but VWF D4 activated pigeon ADAMTS13 ∼4.6-fold. The corresponding selectivity index (∼82) indicates that pigeon ADAMTS13 is ∼1.6-fold more specific for VWF multimers than is human ADAMTS13 (SI ∼54).
Discussion
A common ancestor of ADAMTS13 had a long propeptide and 8 distal T repeats, but many extant lineages have lost the long propeptide and up to 5 of the distal T repeats (supplemental Figure 1). This variability suggests that the MDTCS domains and a few highly conserved distal domains (T7, T8, CUB1, CUB2) could be sufficient for the hemostatic function of ADAMTS13. In agreement with this prediction, deletion of domains T3-T6 from human ADAMTS13 yielded an enzyme that retained allosteric activation on binding to VWF domain D4. In addition, the endogenous signal peptide and propeptide of human or pigeon ADAMTS13 could be replaced with a generic signal peptide without impairing secretion (Figure 3) or allosteric regulation (Table 3). Thus, a fully allosteric ADAMTS13 can be constructed by evolution or by mutagenesis with no propeptide and no more than 3 distal T domains.
Human VWF D4 can bind and allosterically activate ADAMTS13 from many other mammals as well as chicken, pigeon, and X laevis, suggesting the interface between the T7-T8-CUB1 domains and VWF D4 is extraordinarily well conserved. This pattern underscores the importance of binding and allostery for ADAMTS13. For example, binding to VWF increases the local concentration of ADAMTS13, and allosteric activation increases the catalytic efficiency of every bound ADAMTS13, together increasing the efficiency of VWF cleavage by ∼10-fold (Table 3).21,28 Comparable acceleration of VWF cleavage could be achieved without binding or allosteric activation by simply increasing the plasma level of ADAMTS13. However, vertebrates do not appear to use this solution, which suggests that high levels of ADAMTS13 can have adverse consequences. If so, perhaps the allosteric inhibition of unbound ADAMTS13 is useful to prevent the cleavage of other proteins. Fibrinogen has been suggested as 1 such off-target substrate,29 although the evidence has been disputed.30,31
The combination of ADAMTS13 binding to VWF and allosteric activation increases the biological activity of ADAMTS13 in vivo by approximately 1 order of magnitude,21,28 and defects in allosteric activation could be relevant to the pathogenesis of TTP. Approximately 5% to 10% of wild-type ADAMTS13 is enough in humans to prevent thrombotic microangiopathy in most circumstances. However, mutations in ADAMTS13 or VWF that prevent allosteric activation, or auto-Abs with the same effect, might increase the threshold of ADAMTS13 activity required to prevent TTP. In that case, a patient could have the clinical features of TTP with severe ADAMTS13 deficiency, despite having unexpectedly high levels of plasma ADAMTS13 activity when assayed with peptide substrates in vitro.
ADAMTS13 from several species cleaved a bovine VWF71 substrate much more rapidly than their own VWF sequence (Figure 2; Table 2). In addition, systematic mutagenesis of human VWF has identified many amino acid substitutions that impair cleavage by human ADAMTS13, and several others that increase cleavage.32 These findings may be a natural consequence of evolutionary pressure on the ADAMTS13-VWF relationship to balance the risks for VWF-dependent bleeding and thrombosis. Because the risk for microvascular thrombosis does not depend strongly on the level of ADAMTS13 activity, random mutations that cause small decreases in the efficiency of VWF cleavage can be tolerated. Mutations that have slightly more deleterious effects on VWF cleavage might be compensated by increasing the level of circulating ADAMTS13 or by improving the efficiency of allosteric activation. Competition between these mechanisms is likely to account for the observation that ADAMTS13 levels are higher than necessary, and that VWF generally is not an optimal ADAMTS13 substrate.
Presented in abstract form at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3-6 December 2016.
Nucleotide sequences have been submitted to the DDBJ/GenBank/EBI Data Bank with accession numbers BK009996-BK010201. For other original data, please contact pike@wustl.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sharon Deem and colleagues at the St. Louis Zoo; Richard W. Truman and Maria T. Pena at Laboratory Research Branch, National Hansen’s Disease program; and Thomas B. McFadden and colleagues at the Division of Animal Sciences, University of Missouri for plasma sample donations. Also, the authors thank David Ginsburg (University of Michigan) and Linda Pike (Washington University) for their critical reading of this manuscript.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL89746, R01 HL143794, and U54 HL112303 (J.E.S.), a Scientist Development grant from the American Heart Association (16SDG30740011) (J.M.), and a grant from KU Leuven (OT/14/071) (K.V.).
Authorship
Contribution: J.M., J.Z., and J.E.S. conceived and designed experiments; J.M., J.Z., and G.G. performed experiments; J.M., J.Z., G.G., and J.E.S. analyzed data; S.C.G., K.V., and L.A.W. contributed to reagents and materials; J.Z, S.C.G., K.V., G.G., and L.A.W. critically reviewed the manuscript; and J.M. and J.E.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joshua Muia, Department of Medicine, Washington University School of Medicine, 660 S. Euclid Ave, Box 8125, St. Louis, MO 63110; e-mail: jmuia@wustl.edu.