TO THE EDITOR:
Chronic granulomatous disease (CGD), an inherited immunodeficiency resulting from defects in the reduced NAD phosphate oxidase complex rendering phagocytes deficient in producing the superoxide anion necessary for normal killing of bacterial and fungal microorganisms, leads to severe and recurrent infections, inflammatory granulomatous complications, and autoimmune diseases.1 It is associated with substantial morbidity and premature death. Hematopoietic cell transplantation (HCT) is the only long-term curative therapy; however, historically, utility was limited by high rates of graft failure and transplant-related morbidity and mortality. Transplant survival and graft outcome have improved dramatically over the past 10 years as a result of reduced-toxicity conditioning (RTC) regimens, detailed graft-selection hierarchy, superior HLA-matching technology, better cell-dosed grafts, greater availability of grafts, improved supportive care, and more effective antimicrobial therapy. We studied transplant survival post-HCT for CGD in a supraregional immunology transplant center in the United Kingdom.
We report outcomes of patients with CGD who received their first HCT between 1998 and 2017 at the Great North Children’s Hospital, 1 of 2 supraregional transplant centers in the United Kingdom for children with primary immunodeficiency. Outcomes of interest were overall survival (OS), event-free survival (EFS), hematopoietic recovery, toxicities, graft-versus-host disease (GvHD), long-term survival, graft function, posttransplant immune reconstitution, and autoimmune disease posttransplant. OS was defined as survival from first HCT to last follow-up or death. An event was defined as death or second procedure for decreasing chimerism. Cox proportional-hazard modeling was used to analyze predictors of OS and EFS. Variables included for predictor analysis were year of transplant (1998-2007 vs 2008-2017), age at transplant, donor (family vs unrelated donor), conditioning (myeloablative conditioning [MAC] vs RTC), stem cell source (marrow vs peripheral blood), total nucleated cell dose (TNC), and CD34 cell dose. Multiple linear-regression modeling was used to analyze the impact of conditioning, stem cell source, and cell doses on long-term graft function after first successful HCT. CD3, CD4, CD8, CD19, CD27/CD45RA, and CD16/56 enumeration was measured routinely. All data analyses were performed using STATA version 14.2.
Fifty-five children with CGD were included; none were excluded. A detailed description of transplant characteristics is summarized in Table 1. Median age at transplant was 5.3 years (range, 0.6-18.0). Forty-five (82%) had X-linked CGD, and 10 (18%) had autosomal-recessive CGD. Twelve (22%) had growth failure (defined as weight less than the fifth centile for age and sex) at the time of transplant. Of 34 who underwent colonoscopy, 31 (91%) had biopsy-proven colitis. Four (7%) had biopsy-proven granulomatous lung inflammation. One had undefined acute necrotizing encephalitis. Prior to 2007, various conditioning regimens were used, with 21 (38%) patients receiving pharmacokinetically guided IV busulfan and IV cyclophosphamide, with or without serotherapy. From 2007 on, the conditioning regimen was switched to fludarabine–treosulfan–alemtuzumab, with post-HCT GvHD prophylaxis comprising cyclosporine (CSA) and mycophenolate mofetil (MMF) for family and unrelated donors (n = 24; 44%). Fludarabine–treosulfan–thiotepa–anti-thymocyte globulin–rituximab was used for CD3 TCRαβ CD19–depleted haploidentical grafts (n = 4; 7%). Of these 4 patients, 1 received CSA/MMF, and the remaining 3 did not receive any post-HSCT GvHD prophylaxis. The median day to neutrophil and platelet engraftment was 16 days (range, 9-37) and 18 days (range, 10-139), respectively. Ten (20%) patients had grade II-IV acute GvHD, and 5 (9%) patients had grade III-IV acute GvHD. None had chronic GvHD. Only 1 patient who received busulfan-based conditioning had veno-occlusive disease. Eleven (20%) had cytomegalovirus viremia, 5 (9%) had adenoviremia, and 5 (9%) had Epstein-Barr virus viremia.
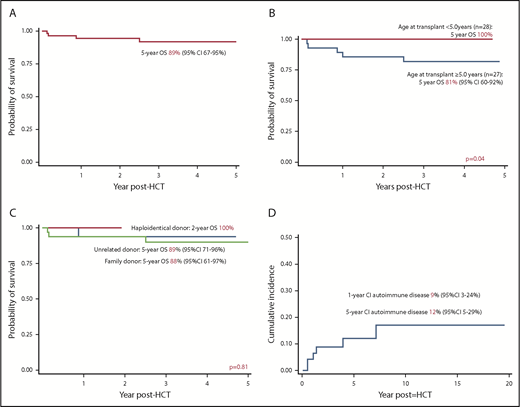
The 5-year OS was 89% (95% confidence interval [CI], 67-95%), increasing to 100% for children transplanted at ≤5 years of age (n = 28) vs 81% (95% CI, 60-92%) for children transplanted at >5 years of age (n = 27; P = .04) (Figure 1A-B). The OS was comparable between matched family (88%; 95% CI, 61-97%) and unrelated donor transplants (89%; 95% CI, 71-95%) (Figure 1C). The 4 haploidentical transplants were successful. Year of transplant (P = .13), conditioning (P = .58), stem cell source (P = .57), TNC (P = .69), and CD34 cell dose (P = .52) were not associated with OS. All 5 deaths were due to transplant-related complications. The median age at transplant for deceased patients was 10.0 years (range, 8.4-18). The 5-year EFS for the cohort was 77% (95% CI, 62-87%). No variables were associated with EFS. All 7 patients with decreasing chimerism received successful second procedures (3 had unconditioned stem cell boost infusion and 4 had fully conditioned second transplant).

Outcomes after HCT for CGD. (A) OS of entire cohort; (B) OS according to age at transplant; (C) OS according to donor type; (D) cumulative incidence of autoimmune disease after HCT for CGD.
Outcomes after HCT for CGD. (A) OS of entire cohort; (B) OS according to age at transplant; (C) OS according to donor type; (D) cumulative incidence of autoimmune disease after HCT for CGD.
The median age of long-term survivors was 14 years (range, 2-36), with a median duration of follow-up of 6.5 years (range, 0.32-19.5). There were no late deaths. None had clinical evidence of colitis. Of the 11 survivors who were >21 years of age, 6 (55%) had unassisted successful pregnancy or fathered children (4 received busulfan-cyclophosphamide, 1 received busulfan-fludarabine, and 1 received fludarabine-melphalan). Median donor myeloid chimerism after first successful HCT (n = 43) was 92% (range, 30-100%). MAC was the only variable associated with higher donor myeloid chimerism (P = .036). MAC (P = .002) and TNC (P = .03) were associated with higher donor T-lymphocyte chimerism (supplemental Figure 1; supplemental Table 1 available on the Blood Web site). For patients who received a second procedure with unconditioned stem cell infusion (n = 3), the median myeloid chimerism was 52% (range, 30-100%), and the median T-lymphocyte chimerism was 26% (range, 16-100%). For patients who received a conditioned second transplant (n = 4), the median myeloid chimerism was higher (89%; range, 30-100%), and the median T-lymphocyte chimerism was 65% (range, 26-100%). Longitudinal immune-reconstitution results were available for 42 patients after first successful HCT (supplemental Figure 2).
The 1- and 5-year cumulative incidence of autoimmune diseases was 9% and 12%, respectively, significantly less than that reported in a recent study.2 Three (5%) had immune cytopenia in the first year post-HCT, whereas 3 (5%) had autoimmune endocrinopathy (2 with thyroid dysfunction; 1 with type 1 diabetes mellitus). Two patients with immune hemolysis achieved remission with IV immunoglobulin and steroid, whereas 1 patient with immune hemolysis and thrombocytopenia needed additional rituximab. None had evidence of immune cytopenia at last follow-up. Autoimmune endocrinopathy occurred 1 year post-HCT (median, 3.9 years; range, 1.4-7.1). One patient with hypothyroidism received thyroxine replacement, 1 patient with Grave’s disease was treated with radioiodine, and 1 patient with type 1 diabetes mellitus received insulin.
The ability of donor-derived neutrophils to replace the recipient’s defective neutrophils makes HCT a superior therapy compared with conventional standard of care using antimicrobial therapy. Previous studies demonstrated that nontransplanted children have more serious infections, more episodes of surgery, poorer growth, and reduced quality of life compared with transplanted children.3-5 Estimated survival of nontransplanted patients was 88% at age 10 years and 55% at age 30 years.6 In this study, our findings emphasize that HCT performed in an experienced immunology transplant center is safe and provides a long-term cure for children with CGD. Alternative donors (unrelated and parental haploidentical donors) are associated with an excellent survival that is comparable to family donors. X-linked carrier family donors should be avoided because they have inflammatory and autoimmune symptoms and excessive fatigue.7-9 Because young age at HCT is associated with a favorable outcome, HCT should be performed as early as possible before the onset of disease-related organ damage. In our center, children with newly diagnosed CGD and neonates that are diagnosed at birth based on family history are recommended for HCT with a matched donor or a haploidentical donor if no suitable matched donor is available. Family screening plays an important role in advancing the transplant care for children with CGD.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all patients and their families, as well as the treating multidisciplinary team.
Authorship
Contribution: S.H.L. collected the data, performed the statistical analysis, interpreted the data, and prepared the manuscript; P.M., H.W., and N.C. collected the data; M.S. and A.R.G. contributed equally to the conceptualization of the research, statistical analysis, interpretation of the data, manuscript writing, and critical review at every stage of the research; and S.O., T.F., S.H., A.C., and M.A. critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Su Han Lum, Great North Children’s Hospital, Victoria Wing, Royal Victoria Infirmary, Newcastle upon Tyne, NE1 4LP, United Kingdom; e-mail: suhanlum@gmail.com.