

TO THE EDITOR:
Recent advances in gene editing technologies using CRISPR/Cas9 allow precise genome editing at a site of interest and have accelerated human disease modeling and the development of corrective gene therapies for various genetic disorders.1,2 We adapted CRISPR/Cas9 editing of rhesus macaque (RM) hematopoietic stem and progenitor cells (HSPCs) to create the first engineered large animal model of a hematologic disease based on close phylogenetic/functional similarity of RM to human HSPCs.3
Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal hematopoietic disorder characterized by intravascular hemolysis, thrombosis, and often bone marrow failure, linked to acquired somatic loss-of-function mutations in HSPCs inactivating the X-linked PIG-A gene that encodes an enzyme necessary for cell surface expression of glycosylphophatidylinositol-anchored proteins (GPI-APs), such as CD55 and CD59.4,5 Mutant HSPCs in PNH clonally expand, often dominating erythroid, myeloid, and B-cell lineages.6 Several hypotheses have been proposed to explain PNH HSPC clonal expansion, including intrinsic resistance to apoptosis,7 additional events such as a second mutation,8 and relative protection of PIG-A–deficient HSPCs from immune attack.4,9,10 However, definitive mechanisms remain unclear. There have been attempts to model PNH by using conditional knockout strategies in mice,11,12 but these do not recapitulate the hemolytic phenotype or clonal dominance.
To provide further insights into PNH, we used CRISPR/Cas9 gene editing of RM HSPCs to create loss-of-function insertions/deletions (indels) in the PIG-A gene, followed by autologous transplantation and long-term follow-up (see supplemental Methods, available on the Blood Web site). We designed 3 guide RNA (gRNA) sequences targeting sites within the PIG-A exon 2 locus on the X chromosome, the most common area for mutations in PNH patients (Figure 1A). All efficiently knocked out GPI-anchored CD55 and CD59 expression in vitro in RM FRhK-4 cells and in CD34+ HSPCs. Peripheral blood HSPCs were mobilized and collected from 1 male (ZL19-M) and 1 female (ZI35-F) RM as described.13,14 Purified CD34+ cells were electroporated with Cas9 protein–gRNA complexes15 and reinfused into the autologous macaque after myeloablative total body irradiation. Each animal received a fraction of CD34+ cells electroporated with the gRNAs targeting PIG-A and a fraction with gRNAs targeting the AAVS1 locus as an internal control for editing at a safe harbor biologically inert site16 (Figure 1A). We confirmed the presence of indels in the infusion product in AAVS1 at similar levels in both macaques, whereas the PIG-A indel frequency was higher in ZL19-M than in ZI35-F, with overall levels lower than at the AAVS1 locus (Figure 1B). Figure 1C summarizes transplantation parameters. After engraftment, the PNH phenotype was assessed via staining with fluorescein-labeled proaerolysin (FLAER), a fluorescent compound that binds to all GPI anchors, and by staining for lineage-specific markers with a focus on granulocytes, which best reflect ongoing HSPC activity and have been shown to correlate with clinical PNH outcomes. Deep sequencing was used to quantitate indels at the targeted sites.
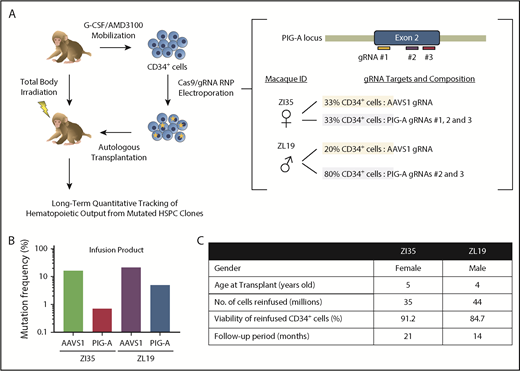
CRISPR/Cas9 gene editing and autologous transplantation of RMs. (A) Schematic diagram of an RM CRISPR/Cas9 gene editing/autologous transplantation model. RM HSPCs were mobilized into the blood with granulocyte colony-stimulating factor (G-CSF) and plerixafor (AMD3100). CD34+ cells were electroporated with ribonucleoprotein (RNP) complexes containing gRNAs targeting either a PIG-A exon or the AAVS1 safe harbor site and Cas9 protein. Edited cells were reinfused into the autologous macaque after total body irradiation. Long-term tracking of mutated cells was performed by targeted deep sequencing and flow cytometry. (B) Before transplantation, initial editing efficiency of PIG-A and AAVS1 in an infusion product was determined in both RMs by targeted deep sequencing. (C) Transplantation characteristics of RMs included in the study.
CRISPR/Cas9 gene editing and autologous transplantation of RMs. (A) Schematic diagram of an RM CRISPR/Cas9 gene editing/autologous transplantation model. RM HSPCs were mobilized into the blood with granulocyte colony-stimulating factor (G-CSF) and plerixafor (AMD3100). CD34+ cells were electroporated with ribonucleoprotein (RNP) complexes containing gRNAs targeting either a PIG-A exon or the AAVS1 safe harbor site and Cas9 protein. Edited cells were reinfused into the autologous macaque after total body irradiation. Long-term tracking of mutated cells was performed by targeted deep sequencing and flow cytometry. (B) Before transplantation, initial editing efficiency of PIG-A and AAVS1 in an infusion product was determined in both RMs by targeted deep sequencing. (C) Transplantation characteristics of RMs included in the study.
Neutrophils and monocytes could be detected with a PNH loss-of-function phenotype at stable levels of 0.2% to 0.6% for more than 2 years, as assessed by loss of FLAER staining and loss of expression of lineage-specific GPI-APs (CD24 on granulocytes and CD14 on monocytes) (Figure 2A; supplemental Figure 1). There was no evidence for intrinsic clonal expansion of GPI– cells over this time period. Targeted deep sequencing confirmed 0.4% to 1% indels at the gRNA-targeted sites in the PIG-A gene. The predominant indel types were single base deletions at the +1 or −1 positions relative to the predicted Cas9 cut sites, but at least 28 indels were identified in the 2 animals and were retrieved from granulocytes and other mature hematopoietic cells stable over time (Figure 2B; supplemental Figure 2). In addition, AAVS1 indels were found at levels of 1.5% to 4.5%, also stable over time (supplemental Figure 3). Given the limited diversity of indels generated by CRISPR/Cas-induced nonhomologous end joining, as evidenced by the sharing of 16 indel types between the 2 animals, we cannot use shared indels between lineages to prove editing of long-term multilineage hematopoietic stem cells, particularly given our data demonstrating that thousands of individual HSPCs contribute to hematopoiesis in our macaque model.17
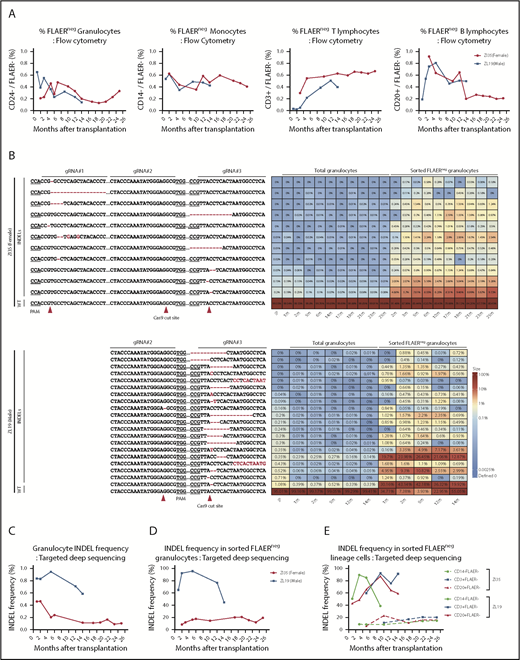
Long-term quantitative tracking of PIG-A–mutated clones by flow cytometry and deep sequencing. (A) Fraction of FLAER– granulocytes, monocytes, T cells, and B cells over time posttransplantation in ZI35 and ZL19 monitored by flow cytometry. (B) Deep sequencing of gRNA target sites within PIG-A exon 2 was performed to quantitate indel frequency and identify individual indel types in total granulocytes and sorted FLAER– granulocytes. Heatmap shows the actual sequences retrieved from ZI35 (top) and ZL19 (bottom), with each row representing a different allele and each column representing a time point. The fractional contribution of individual indels to each sample is represented as a color gradient. Indels are indicated by a red font, and expected Cas9 cut site and protospacer adjacent motif (PAM) are shown in the bottom wild-type (WT) sequence. The fraction of alleles with indels in the 20-bp window surrounding the gRNA target site at the PIG-A exon 2 in total granulocytes (C) and sorted FLAER– granulocytes (D) from ZI35 and ZL19 plotted over time. (E) Targeted deep sequencing of PIG-A exon 2 in ZI35 and ZL19 was performed in FLAER– blood cells from the following lineages over time; monocytes, CD3+ T cells, and CD20+ B cells. The percentage of indels at the target site is shown. IP, infusion product.
Long-term quantitative tracking of PIG-A–mutated clones by flow cytometry and deep sequencing. (A) Fraction of FLAER– granulocytes, monocytes, T cells, and B cells over time posttransplantation in ZI35 and ZL19 monitored by flow cytometry. (B) Deep sequencing of gRNA target sites within PIG-A exon 2 was performed to quantitate indel frequency and identify individual indel types in total granulocytes and sorted FLAER– granulocytes. Heatmap shows the actual sequences retrieved from ZI35 (top) and ZL19 (bottom), with each row representing a different allele and each column representing a time point. The fractional contribution of individual indels to each sample is represented as a color gradient. Indels are indicated by a red font, and expected Cas9 cut site and protospacer adjacent motif (PAM) are shown in the bottom wild-type (WT) sequence. The fraction of alleles with indels in the 20-bp window surrounding the gRNA target site at the PIG-A exon 2 in total granulocytes (C) and sorted FLAER– granulocytes (D) from ZI35 and ZL19 plotted over time. (E) Targeted deep sequencing of PIG-A exon 2 in ZI35 and ZL19 was performed in FLAER– blood cells from the following lineages over time; monocytes, CD3+ T cells, and CD20+ B cells. The percentage of indels at the target site is shown. IP, infusion product.
Sequencing of sorted FLAER– cells greatly increased the indel frequency, confirming that these were valid indels and not sequencing artifacts and that they were responsible for the GPI– phenotype (Figure 2B-D). Note that the expected indel frequency in PIG-A–deficient sorted male FLAER– ZL19-M cells with a single PIG-A allele on the X chromosome would be 100%; however, larger deletions or rearrangements are not detected by standard deep sequencing methodologies,18 and there were likely some differences in sort purity of the rare FLAER– cells, which explains values of <100% and differences between samples sorted independently. The mutation frequencies in total granulocytes and, more importantly, in sorted FLAER– cells in different lineages were always much lower in female ZI35-F cells (Figure 2B-E; supplemental Figure 2). An edited allele frequency in FLAER– female cells half that seen in FLAER– male cells would suggest that only the active X PIG-A allele was edited in female cells. Perhaps X-inactivated alleles offer poor accessibility to editing machinery because of condensed heterochromatin, and this issue warrants further study.
PIG-A mutations have been detected in normal humans at very low levels of 22 per million neutrophils19 and 17 per million T lymphocytes.20 Aerolysin is a toxin that enters cells via GPI-anchored receptors, and aerolysin-resistant colony-forming units were detected at a similarly low frequency; however, unlike PNH patients studied concurrently, healthy individuals had PIG-A mutations that were polyclonal, and in this study, they were not present in T cells, which led the authors to suggest that the mutations were not in hematopoietic stem cells.21 Neither study followed the level of mutant cells over time to address clonal expansion. Rosti et al22 showed stable long-term contributions for up to 13 months of CD24– erythrocytes in chimeric mice derived from pig-a–inactivated embryonic stem cells.
Our data to date suggest that PIG-A mutations in long-term engrafting HSPCs are insufficient to result in rapid intrinsic clonal expansion or a PNH disease phenotype in the macaque model. Clinical parameters related to hemolytic symptoms such as lactic acid dehydrogenase (LDH) remained within the normal range (supplemental Figure 4), which is not surprising given the low PNH clone fraction. Firm conclusions are limited because we analyzed only 2 animals. However, most humans with PNH harbor only a single clone, as documented by sequencing, and in contrast, we followed at least 28 clones between the 2 animals, providing additional power to detect expansion events. Longitudinal human PNH studies show heterogeneous clonal behavior between individuals, with only a minority of clones expanding over time periods of 1 to 6 years,23 suggesting that stochastic or extrinsic factors result in variable clonal behavior. Additional factors are likely necessary, either a second mutational hit, which would cause the further intrinsic selective pressure in favor of the PNH clone, or selective immune-mediated destruction of GPI+ HSPCs in the setting of bone marrow failure.6 Therefore, targeted correction of PIG-A–mutated PNH HSPCs would be unlikely to reverse the PNH phenotype, given the lack of intrinsic clonal expansion of these cells and, in fact, could expose corrected cells to further autoimmune attack if expansion of PNH cells occurs via an immune escape mechanism. Longer follow-up and additional animals will help answer these questions. This model could be further used to investigate extrinsic or intrinsic factors responsible for clonal expansion in a larger cohort of animals with longer follow-up. Our model represents the first NHP gene editing model created to investigate the pathophysiology of a human disease.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health, DNA Sequencing and Genomics and Flow Cytometry Cores, Keyvan Keyvanfar, Stephanie Sellers, and Lemlem Alemu for technical assistance, Bruce E. Hayward and Karen Usdin for constructive comments, and the NHLBI Primate Facility staff.
This work was supported by the Korean Visiting Scientist Training Award (T.-H.S. and K.-R.Y.), and the Intramural Research Program of the NHLBI.
Authorship
Contribution: T.-H.S., E.J.B., K.-R.Y., J.-Y.M., and C.E.D. conceived the study; S.C. and A.A.A. analyzed and processed the data; T.-H.S., E.J.B., K.-R.Y., and C.E.D. interpreted the data; R.E.D. provided technical support; T.-H.S., E.J.B., M.A.F.C., J.-Y.M., A.A.A., S.C., Y.Z., and K.-R.Y. performed experiments; C.E.D. supervised the study; and T.-H.S., K.-R.Y., and C.E.D. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kyung-Rok Yu, Department of Life Sciences, College of Medicine, The Catholic University of Korea, 222 Banpo-daero Seocho-gu, Seoul 06591, Republic of Korea; e-mail: kryu@catholic.ac.kr; and Cynthia E. Dunbar, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Dr, Building 10-CRC, 4-5132, MSC-1202, Bethesda, MD 20892; e-mail: dunbarc@nhlbi.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal