Key Points
CD8+ T cells from CLL patients display aberrations in mitochondrial and glucose metabolism prior to and after stimulation.
CD8+ CD19-CAR T cells have enhanced mitochondrial biogenesis in complete responding CLL patients correlating to expansion and persistence.

Abstract
In chronic lymphocytic leukemia (CLL), acquired T-cell dysfunction impedes development of effective immunotherapeutic strategies, through as-yet unresolved mechanisms. We have previously shown that CD8+ T cells in CLL exhibit impaired activation and reduced glucose uptake after stimulation. CD8+ T cells in CLL patients are chronically exposed to leukemic B cells, which potentially impacts metabolic homeostasis resulting in aberrant metabolic reprogramming upon stimulation. Here, we report that resting CD8+ T cells in CLL have reduced intracellular glucose transporter 1 (GLUT1) reserves, and have an altered mitochondrial metabolic profile as displayed by increased mitochondrial respiration, membrane potential, and levels of reactive oxygen species. This coincided with decreased levels of peroxisome proliferator-activated receptor γ coactivator 1-α, and in line with that, CLL-derived CD8+ T cells showed impaired mitochondrial biogenesis upon stimulation. In search of a therapeutic correlate of these findings, we analyzed mitochondrial biogenesis in CD19-directed chimeric antigen receptor (CAR) CD8+ T cells prior to infusion in CLL patients (who were enrolled in NCT01747486 and NCT01029366 [https://clinicaltrials.gov]). Interestingly, in cases with a subsequent complete response, the infused CD8+ CAR T cells had increased mitochondrial mass compared with nonresponders, which positively correlated with the expansion and persistence of CAR T cells. Our findings demonstrate that GLUT1 reserves and mitochondrial fitness of CD8+ T cells are impaired in CLL. Therefore, boosting mitochondrial biogenesis in CAR T cells might improve the efficacy of CAR T-cell therapy and other emerging cellular immunotherapies.
Introduction
The therapeutic possibilities for chronic lymphocytic leukemia (CLL) have greatly increased over the last few years. Novel agents such as ibrutinib and venetoclax induce high response rates and are generally well tolerated, but their use as monotherapeutic agents is not curative. As a consequence, continuous therapy is required, leading not only to long-lasting remissions1,2 but also to high costs, toxicity, lower compliance, and an increased risk of resistance. Indeed, for both drugs, mechanisms of resistance have now been described that are directly attributable to long-term drug exposure.3,4 Promising results are obtained with novel agents in combination strategies allowing for long-lasting treatment-free responses, but are at this point not expected to be curative.5,6 Therefore, an unmet need exists for the development of additional effective yet tolerable treatment options with alternative mechanisms of action. In contrast to the aforementioned approaches, T-cell–mediated therapy has promising potential in CLL.7-10 Current autologous T-cell–based therapies, such as immune checkpoint inhibition and chimeric antigen receptor (CAR) T cells yield remarkable responses in some patients with advanced relapsed/refractory (R/R) CLL, but only in the minority of patients.11-16 Results of recent trials indicate that CAR T-cell therapy has the potential of inducing sustained remissions in CLL, but does so only in one-third of patients.14 However, the underlying reason for this poor response is unknown.
A likely factor in the limited responses of CAR T-cell therapy in CLL is the acquired T-cell dysfunction that progresses throughout the disease.17-19 T-cell abnormalities include impaired proliferative capacity, an exhaustion phenotype, and diminished CD8+ T-cell cytotoxicity.19-21 CLL patients also display a subset distribution skewed toward an effector memory phenotype, particularly in cytomegalovirus-positive patients.22,23 Increasing evidence suggests that T-cell dysfunction in CLL occurs through direct and indirect interactions of CLL cells with both CD4+ and CD8+ T cells. CLL cells express high levels of inhibitory molecules including programmed death ligand 1, B7-H3, CD270, and the immune-regulatory molecule CD200.24 These molecules have been shown to be key mediators of acquired T-cell synapse defects through CD200R, programmed death 1 (PD-1), and B- and T-lymphocyte attenuator binding to cognate receptors on T cells.21,23,24 Furthermore, molecular and functional defects are also acquired by coculture of previously healthy T cells with CLL cells, implicating a direct immunosuppressive effect by leukemic B cells.20,25,26
Recent studies have shown an intricate relationship between T-cell function and cellular metabolism.27-31 Quiescent T cells primarily use mitochondrial oxidative phosphorylation (OXPHOS) to meet their energy demands. When T cells receive activating signals, a rapid switch to the dominant use of glycolysis takes place.32,33 The conversion to anabolic metabolism is required for full effector function.27 In nutrient-restricted niches, such as in the tumor microenvironment of solid tumors, T cells can become deprived of sufficient amounts of glucose required to execute effector functions.34,35 In CLL, secondary lymphoid organs function as the tumor microenvironment, where T cells are in close contact with CLL cells.36,37 We have previously demonstrated glycolytic impairment in activated CD8+ T cells from CLL patients.38 However, the chronic exposure of CD8+ T cells to leukemic B cells in these patients can potentially impact metabolic homeostasis in resting T cells, which can have consequences for metabolic reprogramming upon stimulation. Because mitochondrial OXPHOS is required for the first steps of T-cell activation upon stimulation,27,39 and for the rapid switch to glycolysis,29 we aimed to determine whether CLL cells impair mitochondrial function. Our findings indicate that CD8+ T cells display a CLL-mediated impairment of mitochondrial biogenesis and fitness, accompanied by reduced glucose transporter 1 (GLUT1) reserves. The translational relevance of these findings was demonstrated by increased mitochondrial biogenesis in CD19-directed CAR+CD8+ T cells in infusion products of CLL patients that show a complete response compared with nonresponders (NRs). Furthermore, mitochondrial biogenesis correlated with the persistence of these cells after infusion and with clinical outcome.
Materials and methods
Patients and HD materials
Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood samples from untreated CLL patients (CD19+/CD5+/CD23+ lymphocytosis and a white blood cell count >20 × 109 cells per liter; Table 1) or age-matched healthy donors (HDs; Table 2; Sanquin, Amsterdam, The Netherlands), and cryopreserved as described previously.22 CAR T-cell infusion products were obtained from CLL patients enrolled in 2 clinical trials of single-agent CTL019 therapy at the University of Pennsylvania (registered at clinicaltrials.gov: NCT01029366, and NCT01747486; Table 3), and generated from R/R CLL patients as previously described.14 Response to CAR T-cell therapy was based on criteria provided by the International Workshop Group on CLL.40 Blood was obtained following written and informed consent. The medical ethical and biobank committees at the Academic Medical Center and the University of Pennsylvania confirmed ethical approval in accordance with the Declaration of Helsinki.
Flow cytometry and cell culture
PBMCs were stained with antibodies (details in supplemental Methods and supplemental Table 1, available on the Blood Web site), measured on an LSR Fortessa (BD Biosciences), and analyzed using FlowJo 10.5.3. CD8+ T cells were positively sorted by using a FacsAriaII, or enriched by using an EasySep Human T Cell Enrichment kit (Stemcell). T cells were analyzed either directly ex vivo, or stimulated using CD3 (clone 1XE) and CD28 (clone 15E8) antibodies for 2 to 5 days.
Extracellular flux analysis
Seahorse XF96 or XFp extracellular flux analyzers (Agilent) were used to analyze sorted CD8+ T cells, as previously described.41 Spare respiratory capacity (SRC) was calculated as the ratio between maximum and basal oxygen consumption rates (OCR). Results were analyzed using Seahorse Wave version 2.4.
Gene-expression profiling
A publicly available microarray data set was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG)-pathway analysis (GSE8835),25 using the statistical software package R2. Mitochondrial (ND1) and nuclear (β-globulin) DNA, and GLUT1 messenger RNA (mRNA) were measured by quantitative polymerase chain reaction (qPCR) using a StepOnePlus Real-Time PCR system, or a Quantstudio 3 (Applied Biosystems/Thermo Fischer Scientific). Data were analyzed using StepOne v2.1, or Quantstudio Design&Analysis v1.4.3.
Glucose and lactate quantification
Glucose and lactate in supernatants were measured turbidimetrically on a Cobas8000 (Roche).
Data presentation and statistical analysis
Flow analysis data obtained from multiple independent experiments were pooled and shown as percentages of positive cells. Data measured within 1 experiment are plotted as mean fluorescence intensity (MFI). For some experiments, both percentages and MFI are shown. Normality of distribution of data were determined by performing a D’Agostino-Pearson omnibus test. For nonparametric distributions, P values were calculated by a Mann-Whitney U test for unpaired data, and the Wilcoxon T test for paired data. For parametric and unpaired data, a Student t test with a Welch correction was used if unequal variance was significant as determined by an F test. A paired Student t test was used for parametric distributed paired data. Pearson correlation coefficients were calculated to determine correlations between 2 pairs of data sets. All statistical tests were 2-tailed and performed using Graphpad PRISM version 7.03. Differences were considered statistically significant if P < .05.
Results
Impaired activation and reduced glucose uptake by CLL-derived CD8+ T cells upon stimulation
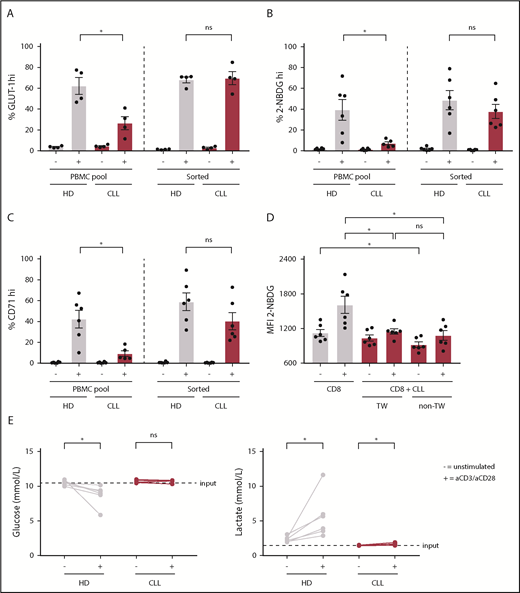
To compare the response of CD8+ T cells derived from untreated CLL patients to age-matched HDs to T-cell receptor triggering, we stimulated PBMCs with anti-CD3/CD28 antibodies. Following stimulation, CLL-derived CD8+ T cells displayed reduced expression of the activation markers CD25, CD38, and CD71 (Figure 1A; supplemental Figure 1A), as well as the degranulation marker CD107a (lysosome-associated membrane protein 1) as compared with HDs (Figure 1B). In addition, the frequency of PD-1+ cells before and after stimulation was significantly higher in CLL-derived CD8+ T cells compared with HDs (Figure 1C; supplemental Figure 1B). As previously reported,21 interferon-γ and tumor necrosis factor-α production was not impaired (supplemental Figure 1C), whereas proliferation was diminished in CLL-derived CD8+ T cells (supplemental Figure 1D). Because adequate T-cell effector function requires a rapid switch to glycolysis,27 and glucose uptake is mediated by expression of the glucose transporter GLUT1,42 we investigated GLUT1 expression by CD8+ T cells in PBMCs 2 days after stimulation with anti-CD3/CD28. Although surface expression of the glucose transporter GLUT1 was comparable between resting CLL and HD CD8+ T cells, CLL-derived CD8+ T cells had lower surface GLUT1 expression after stimulation (Figure 1D; supplemental Figure 1E-F). In line with these data, we found impaired glucose uptake in CLL-derived CD8+ T cells 2 days after stimulation, but not in unstimulated CD8+ T cells (Figure 1E; supplemental Figure 1A). Interestingly, a small group of CLL patients showed similar glucose uptake upon stimulation as in HD T cells, and we compared disease progression (percentage of CLL or white blood cell counts) of the good responders vs poor responders (cutoff: 40% 2-NBDGhi), but we did not observe any significant differences (supplemental Figure 1G). To exclude the possibility that these results were due to different percentages of CD28+ cells or lower expression levels of CD28 per cell between HDs and CLL CD8+ T cells (supplemental Figure 1H), we also stimulated PBMCs from HDs and CLL patients with anti-CD3 alone because CD3 levels were similar in both groups (supplemental Figure 1H).43 We again found impaired GLUT1 surface expression and glucose uptake in CLL-derived CD8+ T cells when stimulated with anti-CD3 alone (supplemental Figure 1I-J). Upon stimulation, CLL-derived CD8+ T cells did not show changes in phosphorylation on AKT on residue S473 (supplemental Figure 2A), phosphatidylinositol 3-kinase (supplemental Figure 2B), or pS6 (supplemental Figure 2C). However, decreased phosphorylation of AKT on residue T308 (supplemental Figure 2D), and 4E-BP1 (indicating reduced translation of proteins) (supplemental Figure 2E) was observed after T-cell stimulation, as well as a clear trend toward reduced expression of hypoxia-inducible factor 1α (HIF-1α) (supplemental Figure 2F). Taken together, these data demonstrate that, upon stimulation, CLL-derived CD8+ T cells have an impaired ability to become activated, and to sufficiently increase their glucose uptake, which is at least in part due to a lower expression of GLUT1 on the cell surface.
![Figure 1. Impaired activation and reduced glucose uptake by CLL-derived CD8+ T cells upon stimulation. PBMCs from CLL patients and age-matched HDs were thawed and cultured for 2 (A-B,D-F) or 5 (C) days in the presence or absence of anti-CD3/CD28 antibodies. Cells were analyzed for (A) activation markers (CLL, n = 15-19; HD, n = 16-22), (B) degranulation (CLL, n = 12; HD, n = 12), (C) PD-1 expression (CLL, n = 8; HD, n = 8), (D) surface glucose transporter GLUT1 (measured by GLUT1 Ras-binding domain [RBD] green fluorescent protein [GFP] construct) (CLL, n = 6; HD, n = 4; data are the same as in supplemental Figure 1G), and (E) glucose uptake (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose [2-NBDG]; CLL, n = 23; HD, n = 20; data are the same as in supplemental Figure 1F), and expression of (F) HIF-1α (CLL, n = 6; HD, n = 4). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by an unpaired Student t test (A-C,E), or a Mann-Whitney test (A,D,F). Data are presented as mean plus or minus standard error of the mean (SEM). *P < .05; **P < .005; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/1/10.1182_blood.2018885863/3/m_blood885863f1.png?Expires=1770145992&Signature=R~39ODMUtexD3VSnBvOii1t96-8KrHDQDWNd-giA7uVly4LSCfMpGyEsh6n54WyD2YMV~3VgGNNf60dMURIM~vkVHXCjSwhiJvPOouKBjscFdp-CAcVdTqYl7a3SIEiT4CxLVn0R7pdhmuRPtGhw4QNmzeNJ0I1q3-WPtPxHaqfYRrWIhmkeURRq701Pmw6g6AGNntrpsCzhzZovUtvHH7Site8FsoK9PoN6n5hzje0oiiWAO7RZc80mDoPc0HCfZi1TFaUdq0QCUhdsfA3S-2KvcaiGFaFCkPU3VkH6-KGy27~BDzzl2XJzzHMzXV2Sutdlz~HkPahuaCT8QS~PXQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Impaired activation and reduced glucose uptake by CLL-derived CD8+ T cells upon stimulation. PBMCs from CLL patients and age-matched HDs were thawed and cultured for 2 (A-B,D-F) or 5 (C) days in the presence or absence of anti-CD3/CD28 antibodies. Cells were analyzed for (A) activation markers (CLL, n = 15-19; HD, n = 16-22), (B) degranulation (CLL, n = 12; HD, n = 12), (C) PD-1 expression (CLL, n = 8; HD, n = 8), (D) surface glucose transporter GLUT1 (measured by GLUT1 Ras-binding domain [RBD] green fluorescent protein [GFP] construct) (CLL, n = 6; HD, n = 4; data are the same as in supplemental Figure 1G), and (E) glucose uptake (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose [2-NBDG]; CLL, n = 23; HD, n = 20; data are the same as in supplemental Figure 1F), and expression of (F) HIF-1α (CLL, n = 6; HD, n = 4). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by an unpaired Student t test (A-C,E), or a Mann-Whitney test (A,D,F). Data are presented as mean plus or minus standard error of the mean (SEM). *P < .05; **P < .005; ****P < .0001.
Impaired activation and reduced glucose uptake by CLL-derived CD8+ T cells upon stimulation. PBMCs from CLL patients and age-matched HDs were thawed and cultured for 2 (A-B,D-F) or 5 (C) days in the presence or absence of anti-CD3/CD28 antibodies. Cells were analyzed for (A) activation markers (CLL, n = 15-19; HD, n = 16-22), (B) degranulation (CLL, n = 12; HD, n = 12), (C) PD-1 expression (CLL, n = 8; HD, n = 8), (D) surface glucose transporter GLUT1 (measured by GLUT1 Ras-binding domain [RBD] green fluorescent protein [GFP] construct) (CLL, n = 6; HD, n = 4; data are the same as in supplemental Figure 1G), and (E) glucose uptake (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose [2-NBDG]; CLL, n = 23; HD, n = 20; data are the same as in supplemental Figure 1F), and expression of (F) HIF-1α (CLL, n = 6; HD, n = 4). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by an unpaired Student t test (A-C,E), or a Mann-Whitney test (A,D,F). Data are presented as mean plus or minus standard error of the mean (SEM). *P < .05; **P < .005; ****P < .0001.
Reduced glucose uptake in CLL-derived CD8+ T cells is reversible and induced by a soluble factor
To investigate whether the metabolic alterations in T cells were imposed by CLL cells, we stimulated purified CD8+ T cells or CD8+ T cells in their PBMC pool, which in our experiments consist of, on average, 88% CLL cells, with anti-CD3/CD28 antibodies. In contrast to CD8+ T cells stimulated in their PBMC pool, purified CLL-derived CD8+ T cells had similar surface GLUT1 expression, glucose uptake, and CD71 expression after stimulation as HD CD8+ T cells (Figure 2A-C). To elucidate whether CLL cells were directly responsible for the dampening effects on activation and glucose uptake of T cells, we performed coculture experiments of autologous purified CD8+ T cells and purified CLL cells, whether separated by a transwell membrane or not. Upon stimulation, CLL-derived CD8+ T cells cultured in the presence of CLL cells showed impaired glucose uptake compared with CLL-derived CD8+ T cells cultured alone, independent of direct cell-cell contact (Figure 2D). These data suggest that in the presence of CLL cells a soluble factor is responsible for the impaired glucose uptake by CD8+ T cells. Because CLL cells might compete with T cells for glucose, as has been shown in a solid tumor model,34 and given the fact that lactate can inhibit glycolysis,34,44,45 we tested whether glucose or lactate concentrations were a limiting factor in PBMC cultures from CLL patients. As expected, glucose levels decreased and lactate levels increased in supernatants from HD PBMCs upon stimulation as compared with unstimulated PBMCs (Figure 2E). In contrast, in PBMC cultures derived from CLL patients, glucose and lactate concentrations did not, or only marginally, change compared with unstimulated PBMCs. This observation suggests that the reduced glucose uptake by CLL-derived CD8+ T cells cannot be explained by reduced glucose availability in the media or by repressive levels of lactate. Together, these data indicate that the suppression of glucose uptake in CD8+ cells from CLL patients upon stimulation is not T-cell intrinsic, but rather a consequence of exposure to CLL cells.
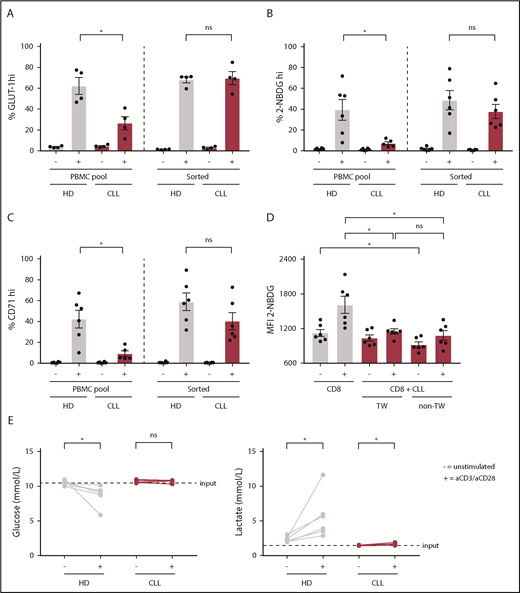
Reduced glucose uptake in CLL-derived CD8+ T cells can be reversed and is not attributable to competition for glucose with CLL cells. CLL-derived CD8+ T cells and age-matched HDs were either positively sorted by fluorescence-activated cell sorting (FACS) or by using a negative T-cell selection kit from Stemcell or left in their PBMC fraction, supplemented with IL-2, and stimulated for 2 days using anti-CD3/CD28 antibodies. After stimulation, cells were analyzed for (A) surface GLUT1 expression (CLL, n = 4; HD, n = 4), (B) glucose uptake (2-NBDG; CLL, n = 5-6; HD, n = 6), and (C) activation (CD71; CLL, n = 5-6; HD, n = 6). (D) CLL cells from PBMCs from CLL patients were purified using CD19 macrophage-activated cell sorting and CD8+ T cells were cell sorted afterward. CD8+ T cells were either cultured alone, or with CLL cells separated by a transwell (0.2 µm pore size), or cultured together with CLL cells without a transwell in a 1:9 ratio, in the presence of IL-2. Two days after stimulation using anti-CD3/CD28 antibodies, CD8+ T cells were analyzed for glucose uptake (2-NBDG) by flow cytometry (CLL, n = 6). (E) Supernatants of PBMCs from CLL patients and age-matched HDs stimulated for 2 days with anti-CD3/CD28 antibodies were analyzed for glucose and lactate concentrations (CLL, n = 6; HD, n = 6). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Mann-Whitney test (A-C), or a Wilcoxon signed rank test (D-E). Data are presented as mean plus or minus SEM. *P < .05. ns, not significant; TW, transwell.
Reduced glucose uptake in CLL-derived CD8+ T cells can be reversed and is not attributable to competition for glucose with CLL cells. CLL-derived CD8+ T cells and age-matched HDs were either positively sorted by fluorescence-activated cell sorting (FACS) or by using a negative T-cell selection kit from Stemcell or left in their PBMC fraction, supplemented with IL-2, and stimulated for 2 days using anti-CD3/CD28 antibodies. After stimulation, cells were analyzed for (A) surface GLUT1 expression (CLL, n = 4; HD, n = 4), (B) glucose uptake (2-NBDG; CLL, n = 5-6; HD, n = 6), and (C) activation (CD71; CLL, n = 5-6; HD, n = 6). (D) CLL cells from PBMCs from CLL patients were purified using CD19 macrophage-activated cell sorting and CD8+ T cells were cell sorted afterward. CD8+ T cells were either cultured alone, or with CLL cells separated by a transwell (0.2 µm pore size), or cultured together with CLL cells without a transwell in a 1:9 ratio, in the presence of IL-2. Two days after stimulation using anti-CD3/CD28 antibodies, CD8+ T cells were analyzed for glucose uptake (2-NBDG) by flow cytometry (CLL, n = 6). (E) Supernatants of PBMCs from CLL patients and age-matched HDs stimulated for 2 days with anti-CD3/CD28 antibodies were analyzed for glucose and lactate concentrations (CLL, n = 6; HD, n = 6). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Mann-Whitney test (A-C), or a Wilcoxon signed rank test (D-E). Data are presented as mean plus or minus SEM. *P < .05. ns, not significant; TW, transwell.
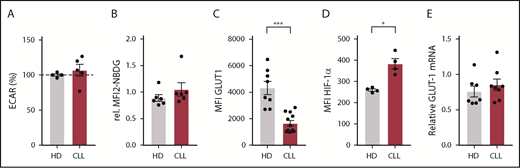
Diminished GLUT1 reserves prior to stimulation
Because the effects of CLL cells on T cells can already have an impact on resting T cells during disease progression, we next focused on the metabolic status of CLL-derived CD8+ T cells prior to stimulation. We found no differences in the extracellular acidification rate (ECAR; Figure 3A) an indicator of glycolysis, between resting CD8+ T cells from CLL and HDs, which was in line with similar surface expression of GLUT1 (Figure 1D) and glucose uptake (Figures 1E and 3B) in unstimulated CD8+ T cells from CLL and HDs. In contrast, total GLUT1 (including intravesicular reserves) in resting CLL-derived CD8+ T cells was diminished (Figure 3C). HIF-1α is the main regulator of GLUT1 transcription,46 and lower intracellular GLUT1 reserves could potentially result from low HIF-1α expression. However, we found higher expression of HIF-1α in CD8+ cells from CLL patients (unstimulated CD8+ T cells; Figures 1F and 3D), and similar GLUT1 mRNA levels in CD8+ cells from CLL patients and HDs (Figure 3E). Together, these data suggest that reduced availability of intracellular GLUT1 reserves prior to stimulation can at least in part explain the reduced surface expression of GLUT1 after stimulation.
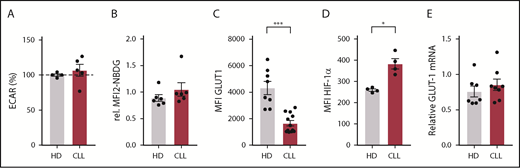
Expression of intracellular GLUT1 is diminished in CLL-derived CD8+ T cells. CD8+ T cells were sorted (A,E) from PBMCs from CLL patients and age-matched HDs or were left in their PBMC fraction (B-D), and analyzed for basal (A) extracellular acidification rate (ECAR; indicating glycolysis; CLL, n = 8; HD, n = 10), (B) glucose uptake, (C) total expression of GLUT1 (intracellular plus surface; CLL, n = 4; HD, n = 4), (D) HIF-1α expression (CLL, n = 4; HD, n = 4), and (E) GLUT-1 mRNA (CLL, n = 8; HD, n = 7). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Welch test (A,C), Mann-Whitney test (B,D), or an unpaired Student t test (E). Data are presented as mean plus or minus SEM. *P < .05.
Expression of intracellular GLUT1 is diminished in CLL-derived CD8+ T cells. CD8+ T cells were sorted (A,E) from PBMCs from CLL patients and age-matched HDs or were left in their PBMC fraction (B-D), and analyzed for basal (A) extracellular acidification rate (ECAR; indicating glycolysis; CLL, n = 8; HD, n = 10), (B) glucose uptake, (C) total expression of GLUT1 (intracellular plus surface; CLL, n = 4; HD, n = 4), (D) HIF-1α expression (CLL, n = 4; HD, n = 4), and (E) GLUT-1 mRNA (CLL, n = 8; HD, n = 7). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Welch test (A,C), Mann-Whitney test (B,D), or an unpaired Student t test (E). Data are presented as mean plus or minus SEM. *P < .05.
Elevated mitochondrial membrane potential and mitochondrial reactive oxygen species levels in CLL-derived CD8+ T cells
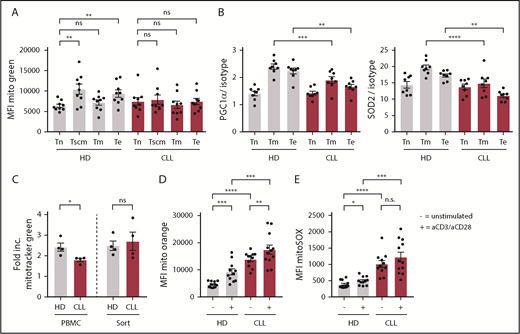
Because mitochondrial metabolism is instrumental in engaging the first steps of glycolysis upon T-cell stimulation by enabling the conversion of glucose into glucose-6-phosphate,29 we next investigated OXPHOS in CD8+ T cells prior to stimulation. Using a publicly available database,25 we analyzed gene-expression profiles of CLL patient and HD-derived CD8+ T cells using KEGG pathway analysis, and found OXPHOS as the second most differentially expressed pathway (Figure 4A). Specifically, genes encoding subunits of the electron transport chain complexes I, III, IV, and V, and the mitochondrial-associated protein phosphatase 2 were expressed at a higher level in CLL-derived CD8+ T cells (Figure 4A). Accordingly, we found increased OXPHOS levels as indicated by higher OCR (Figure 4B; supplemental Figure 3A), and increased adenosine triphosphate production in CLL-derived CD8+ T cells (supplemental Figure 3B). This increase in OXPHOS was not attributable to higher mitochondrial mass as measured by Mitotracker Green staining and the relative levels of mitochondrial DNA (Figure 4C).47 We did not observe any significant differences between HD or CLL-derived CD8+ T cells when analyzing proton leak or maximum respiration (supplemental Figure 3B). The SRC, indicating the capacity of a cell to deal with increased cellular bioenergetic demands such as T-cell activation,28,48 however, was reduced in CLL-derived CD8+ T cells (Figure 4D).
![Figure 4. Altered mitochondrial homeostasis in CLL-derived CD8+ T cells. (A) KEGG pathway analysis showing top 10 of most significantly different pathways in CD8+ T cells from HDs vs CLL, and heatmap of differentially expressed genes in OXPHOS pathway (Gene Expression Omnibus [GEO] accession number GSE8835; P = 3.4 × 10−15 for OXPHOS pathway). PBMCs from CLL patients and age-matched HDs were analyzed by flow cytometry (C,E-H) or Seahorse EFA (B,D) either directly after thawing (B-D,H), 2 days of culturing (E-F), or 3 days of culturing (G). (B) Cells were analyzed for OCR (indicating OXPHOS; CLL, n = 7; HD, n = 6). (C) Mitochondrial mass was determined by flow cytometry (Mitotracker Green; CLL, n = 29; HD, n = 29) and by qPCR by calculating the mitochondrial DNA (mtDNA)-to-nuclear DNA (nDNA) ratio (CLL, n = 8; HD, n = 5). (D) SRC was calculated as the ratio of maximum OCR over basal OCR (CLL, n = 6; HD, n = 6). (E) ΔΨm was determined by flow cytometry (Mitotracker Orange; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5D). (F) Mitochondrial ROS (mitoSOX; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5E). (G) Mitochondrial ROS as well as mitochondrial potential were analyzed in HD CD8 T cells cocultured with HD-derived B cells or CLL (HD, n = 8). (H) PGC-1α, SOD1, and SOD2 (CLL, n = 8; HD, n = 8), HO-1, NRF-2, and ERRα (CLL, n = 8; HD, n = 4). Data were normalized to HD to compile multiple independent experiments (B,D). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Welch test (E-F), Mann-Whitney test (B-D,G-H), or an unpaired Student t test (H). Data are presented as mean plus or minus SEM. *P < .05; **P < .005; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/1/10.1182_blood.2018885863/3/m_blood885863f4.png?Expires=1770145992&Signature=NQTTE9dP3Q4eFZRUl86IUcIgi~lWMBtDAHRQrmBzj262vnzubL6CuT6CVlbSDSTYno8nwc79RtWm3CXKwjmjdt7nLNEb5pFjGgNDeAOC-P7FVWbCFjxXAUrb1pBW2J392ag-R-l2OciskfOzS~1cGIMhq9917UXqsjxYzHEKmg3Y~lHi8cxIANEYLKWYB9CFQnk-4Bkez975R8A4nOClsr-f5B3whZa3Wy04EFlJNG4NWsQokmGAhJfD9GK4bo0o2z5ZzIVG5Tq-TZhVYNobPiXHAMAk~OEj2Wy8RRG0135sbvxU08N1ua9SadEc~8xR7XTPd35zFnK1NvLIvafpQA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Altered mitochondrial homeostasis in CLL-derived CD8+ T cells. (A) KEGG pathway analysis showing top 10 of most significantly different pathways in CD8+ T cells from HDs vs CLL, and heatmap of differentially expressed genes in OXPHOS pathway (Gene Expression Omnibus [GEO] accession number GSE8835; P = 3.4 × 10−15 for OXPHOS pathway). PBMCs from CLL patients and age-matched HDs were analyzed by flow cytometry (C,E-H) or Seahorse EFA (B,D) either directly after thawing (B-D,H), 2 days of culturing (E-F), or 3 days of culturing (G). (B) Cells were analyzed for OCR (indicating OXPHOS; CLL, n = 7; HD, n = 6). (C) Mitochondrial mass was determined by flow cytometry (Mitotracker Green; CLL, n = 29; HD, n = 29) and by qPCR by calculating the mitochondrial DNA (mtDNA)-to-nuclear DNA (nDNA) ratio (CLL, n = 8; HD, n = 5). (D) SRC was calculated as the ratio of maximum OCR over basal OCR (CLL, n = 6; HD, n = 6). (E) ΔΨm was determined by flow cytometry (Mitotracker Orange; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5D). (F) Mitochondrial ROS (mitoSOX; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5E). (G) Mitochondrial ROS as well as mitochondrial potential were analyzed in HD CD8 T cells cocultured with HD-derived B cells or CLL (HD, n = 8). (H) PGC-1α, SOD1, and SOD2 (CLL, n = 8; HD, n = 8), HO-1, NRF-2, and ERRα (CLL, n = 8; HD, n = 4). Data were normalized to HD to compile multiple independent experiments (B,D). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Welch test (E-F), Mann-Whitney test (B-D,G-H), or an unpaired Student t test (H). Data are presented as mean plus or minus SEM. *P < .05; **P < .005; ****P < .0001.
Altered mitochondrial homeostasis in CLL-derived CD8+ T cells. (A) KEGG pathway analysis showing top 10 of most significantly different pathways in CD8+ T cells from HDs vs CLL, and heatmap of differentially expressed genes in OXPHOS pathway (Gene Expression Omnibus [GEO] accession number GSE8835; P = 3.4 × 10−15 for OXPHOS pathway). PBMCs from CLL patients and age-matched HDs were analyzed by flow cytometry (C,E-H) or Seahorse EFA (B,D) either directly after thawing (B-D,H), 2 days of culturing (E-F), or 3 days of culturing (G). (B) Cells were analyzed for OCR (indicating OXPHOS; CLL, n = 7; HD, n = 6). (C) Mitochondrial mass was determined by flow cytometry (Mitotracker Green; CLL, n = 29; HD, n = 29) and by qPCR by calculating the mitochondrial DNA (mtDNA)-to-nuclear DNA (nDNA) ratio (CLL, n = 8; HD, n = 5). (D) SRC was calculated as the ratio of maximum OCR over basal OCR (CLL, n = 6; HD, n = 6). (E) ΔΨm was determined by flow cytometry (Mitotracker Orange; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5D). (F) Mitochondrial ROS (mitoSOX; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5E). (G) Mitochondrial ROS as well as mitochondrial potential were analyzed in HD CD8 T cells cocultured with HD-derived B cells or CLL (HD, n = 8). (H) PGC-1α, SOD1, and SOD2 (CLL, n = 8; HD, n = 8), HO-1, NRF-2, and ERRα (CLL, n = 8; HD, n = 4). Data were normalized to HD to compile multiple independent experiments (B,D). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Welch test (E-F), Mann-Whitney test (B-D,G-H), or an unpaired Student t test (H). Data are presented as mean plus or minus SEM. *P < .05; **P < .005; ****P < .0001.
Mitochondrial membrane potential (ΔΨm) has been shown to be a good indicator of mitochondrial fitness, and low ΔΨm CD8+ T cells display enhanced in vivo persistence, and greater antitumor immunity relative to high ΔΨm cells.49 In line with this, CLL-derived CD8+ T cells showed higher ΔΨm (Figure 4E), which was accompanied by increased mitochondrial reactive oxygen species (ROS) but not total cellular ROS production (Figure 4F; supplemental Figure 3C-D). Additionally, magnetic-activated cell sorted HD-derived CD8+ T cells displayed increased mitochondrial ROS and ΔΨm when cocultured together with CLL cells compared with healthy B cells (Figure 4G), again indicating that CLL cells induce metabolic changes at the mitochondrial level in T cells. Peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) is the main regulator of mitochondrial biogenesis, and the expression of mitochondrial enzymes like superoxide dismutase 2 (SOD2), which is essential in suppressing ROS in the form of O2− generated by mitochondria.50,51 In accordance with increased levels of mitochondrial ROS, we found reduced PGC-1α protein levels in CLL-derived CD8+ T cells, and lower expression of SOD2, but not the cytosolic/intermembrane form SOD1 (Figure 4H). Furthermore, expression of antioxidant heme oxygenase 1 (HO-1), and its regulator nuclear factor erythroid 2–related factor 2 (NRF-2) were lower in CLL-derived CD8+ T cells, whereas expression of estrogen-related receptor α (ERRα) was unchanged (Figure 4H; supplemental Figure 3E). Together, these data indicate that mitochondrial fitness is impaired in CLL-derived CD8+ T cells.
Impaired induction of mitochondrial biogenesis in CLL-derived CD8+ T cells
When healthy T cells become activated, mitochondrial mass increases to be able to deal with enhanced demands of several metabolic needs.52,53 No differences were found in mitochondrial mass between naive CLL- or HD-derived CD8+ T cells (Figure 5A). As expected, antigen-experienced cells displayed increased mitochondrial mass compared with naive CD8+ T cells derived from HDs (Figure 5A). In contrast, in CLL patients, no increase in mitochondrial mass was found in antigen-experienced compared with naive CD8+ T cells (Figure 5A; supplemental Figure 4A). In addition, the reduced expression of PGC-1α and SOD2 in CLL-derived CD8+ T cells was primarily found in antigen-experienced T cells (Figure 5B). These data hint to impaired mitochondrial biogenesis in CLL CD8+ T cells upon T-cell receptor triggering. Indeed, when we stimulated CD8+ T cells in vitro, we found that mitochondrial biogenesis was reduced in CLL-derived CD8+ T cells (supplemental Figure 4B), which could be rescued by depletion of CLL cells (Figure 5C). Moreover, ΔΨm and ROS production was already higher in unstimulated CLL-derived CD8+ T cells compared with HDs (Figure 4E-F), and remained higher after stimulation (Figure 5D-E). Together, these data indicate that CLL-derived CD8+ T cells have impaired mitochondrial biogenesis, which might at least in part explain the hampered activation upon stimulation.
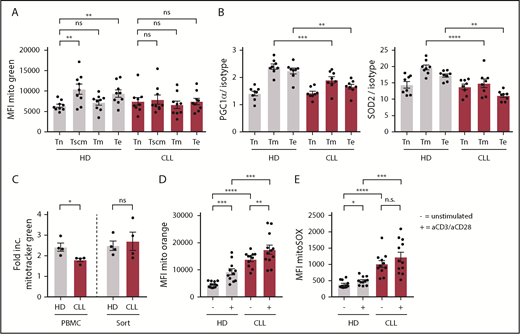
Impaired induction of mitochondrial biogenesis in CLL-derived CD8+ T cells. PBMCs from CLL patients and age-matched HDs were thawed and subsets of CD8+ T cells were directly analyzed by flow cytometry. (A) Mitochondrial mass as defined by Mitotracker Green; naive T (Tn) cells: CD27+CD45RA+CCR7+CD95−; memory stem cells (Tscm): CD27+CD45RA+CCR7+CD95+; memory T cells (Tm): CD27+CD45RA−; and effector T cells (Te): CD27−CD45RA−/+ (CLL, n = 9; HD, n = 9). (B) PGC-1α and SOD2 expression in CD8+ T-cell subsets defined as naive (Tn: CD27+CD45RA+), memory (Tm: CD27+CD45RA−), and effector T cells (Te: CD27−CD45RA−/+; CLL, n = 8; HD, n = 8). (C-E) PBMCs were stimulated by using anti-CD3/CD28 antibodies for 2 days and being analyzed for (C) induction of mitochondrial mass, which was calculated by dividing the MFI of Mitotracker Green of stimulated cells by unstimulated cells (CLL, n = 4; HD, n = 4), (D) ΔΨm (CLL, n = 11-12; HD, n = 11-12; unstimulated control is the same as in Figure 4D), and (E) mitochondrial ROS (CLL, n = 11-12; HD, n = 11-12; unstimulated control is the same as in Figure 4E). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a paired Student t test (A), an unpaired Student t test (B, D), a Mann-Whitney test (C), or a Welch test (D-E). Data are presented as mean plus or minus SEM. *P < .05; **P < .005; ***P < .0005; ****P < .0001.
Impaired induction of mitochondrial biogenesis in CLL-derived CD8+ T cells. PBMCs from CLL patients and age-matched HDs were thawed and subsets of CD8+ T cells were directly analyzed by flow cytometry. (A) Mitochondrial mass as defined by Mitotracker Green; naive T (Tn) cells: CD27+CD45RA+CCR7+CD95−; memory stem cells (Tscm): CD27+CD45RA+CCR7+CD95+; memory T cells (Tm): CD27+CD45RA−; and effector T cells (Te): CD27−CD45RA−/+ (CLL, n = 9; HD, n = 9). (B) PGC-1α and SOD2 expression in CD8+ T-cell subsets defined as naive (Tn: CD27+CD45RA+), memory (Tm: CD27+CD45RA−), and effector T cells (Te: CD27−CD45RA−/+; CLL, n = 8; HD, n = 8). (C-E) PBMCs were stimulated by using anti-CD3/CD28 antibodies for 2 days and being analyzed for (C) induction of mitochondrial mass, which was calculated by dividing the MFI of Mitotracker Green of stimulated cells by unstimulated cells (CLL, n = 4; HD, n = 4), (D) ΔΨm (CLL, n = 11-12; HD, n = 11-12; unstimulated control is the same as in Figure 4D), and (E) mitochondrial ROS (CLL, n = 11-12; HD, n = 11-12; unstimulated control is the same as in Figure 4E). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a paired Student t test (A), an unpaired Student t test (B, D), a Mann-Whitney test (C), or a Welch test (D-E). Data are presented as mean plus or minus SEM. *P < .05; **P < .005; ***P < .0005; ****P < .0001.
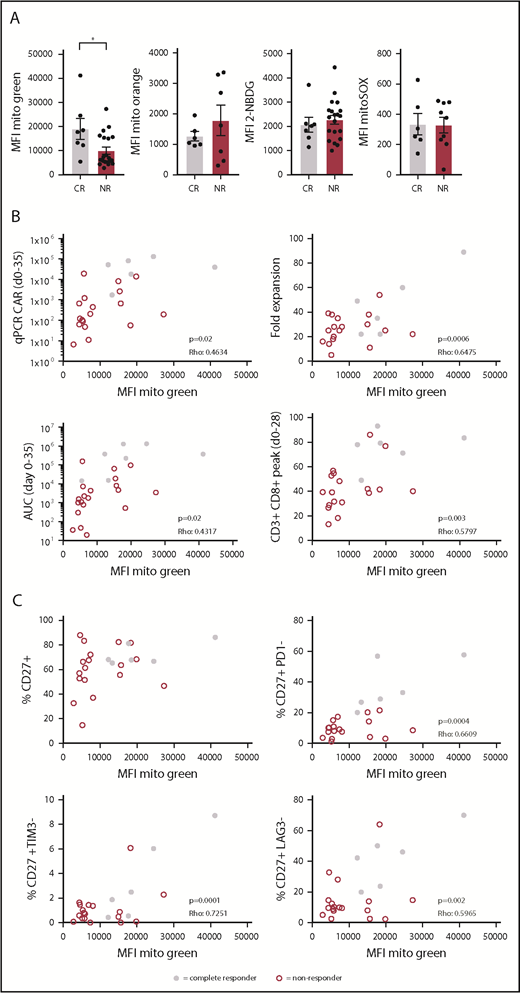
Increased mitochondrial biogenesis in CD19-directed CAR T cells from CLL patients showing a complete response
CAR T-cell therapy is a promising therapy to treat B-cell malignancies such as acute lymphoblastic leukemia and diffuse large B-cell lymphoma. However, only limited successes have been obtained in the treatment of CLL with this approach. We asked whether reduced metabolic fitness of autologous CD8+ T cells could also be observed in transferred CAR T cells to provide a mechanistic basis for the low response rates observed in 2 trials investigating efficacy of CD19 CAR T-cell therapy in CLL patients.14 We analyzed CAR+CD8+ T cells in infusion products of 27 R/R CLL patients enrolled in these CAR T-cell trials for mitochondrial mass, glucose uptake, mitochondrial ROS, and ΔΨm. Although glucose uptake, ΔΨm, and mitochondrial ROS were comparable between complete responders (CRs) and NRs, mitochondrial mass was significantly higher in patients who had a complete response (Figure 6A; supplemental Figure 5). It is important to note that in the process of CAR T-cell generation, T cells are stimulated, thus these data on mitochondrial mass represent mitochondrial biogenesis in response to stimulation. Recent studies provided a strong link between indicators of CAR T-cell persistence and clinical outcome.54-56 A variety of markers used to indicate persistence, including fold expansion of CAR T cells in vitro, expansion peak of CAR T cells at days 0 to 35 using qPCR, area under the curve (AUC) of the CAR T-cell qPCR calculated for days 0 to 35, and peak percentage of CD3+CD8+ cells in the first 28 days postinfusion, indeed showed significant correlations with clinical responses.56 We found positive correlations between mitochondrial mass of CAR+CD8+ T cells in infusion products and those 4 indicators of persistence (Figure 6B). In addition, we observed that mitochondrial mass correlated positively with the presence of nonexhausted CD27+ T cells that were negative for the expression of inhibitory receptors PD-1, T-cell immunoglobulin and mucin-domain containing 3 (TIM-3), and lymphocyte activation gene 3 (LAG-3), but not with presence of CD27+ T cells alone (Figure 6C). Taken together, our data indicate that clinical outcome of R/R CLL patients following CAR T-cell therapy is linked to mitochondrial mass of the infused CAR+CD8+ T cells. Thus, enhancing mitochondrial biogenesis during CAR T-cell production may ultimately lead to more durable clinical responses in CAR T-cell therapy in CLL.
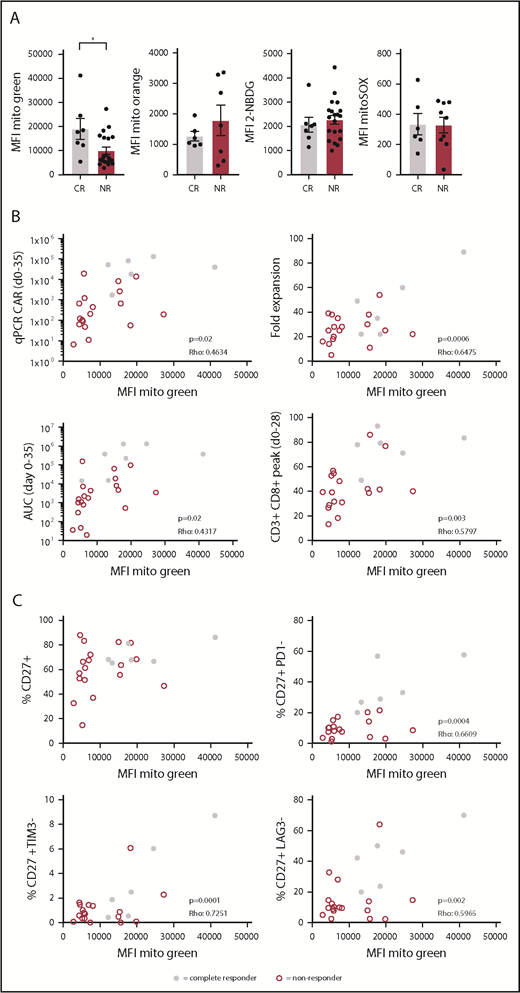
Increased mitochondrial mass in CAR T cells from CLL patients showing complete response to CD19 CAR T-cell therapy. Patients were separated into 2 groups based on response to therapy: CRs and NRs. Viably frozen infusion products from CLL patients undergoing a CD19 CAR T-cell trial (NCT01747486, and NCT01029366; CR, n = 7; NR, n = 20) were thawed and CAR+CD8+ T cells were analyzed for (A) mitochondrial mass (Mitotracker Green), glucose uptake (2-NBDG), mitochondrial membrane potential (Mitotracker Orange), and mitochondrial ROS (MitoSOX). (B) Mitochondrial mass of CAR+CD8+ T cells plotted against determinants of clinical outcome including expansion of the CAR T-cell culture (fold expansion), expansion of the CAR T cells after infusion (qPCR CAR days 0-35), persistence of CAR T cells calculated at days 0 to 35 postinfusion (AUC days 0-35), and overall expansion of CD8+ T cells after infusion (CD3+CD8+ peak days 0-28) (CR, n = 6; NR, n = 18). (C) Mitochondrial mass of CAR+CD8+ T cells plotted against the percentage of CD27+ cells negative for PD-1, TIM-3, and LAG-3 (CR, n = 6; NR, n = 18). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Mann-Whitney test (A). Correlations were determined by a Spearman Rho test (B-C). Data are presented as mean plus or minus SEM. *P < .05.
Increased mitochondrial mass in CAR T cells from CLL patients showing complete response to CD19 CAR T-cell therapy. Patients were separated into 2 groups based on response to therapy: CRs and NRs. Viably frozen infusion products from CLL patients undergoing a CD19 CAR T-cell trial (NCT01747486, and NCT01029366; CR, n = 7; NR, n = 20) were thawed and CAR+CD8+ T cells were analyzed for (A) mitochondrial mass (Mitotracker Green), glucose uptake (2-NBDG), mitochondrial membrane potential (Mitotracker Orange), and mitochondrial ROS (MitoSOX). (B) Mitochondrial mass of CAR+CD8+ T cells plotted against determinants of clinical outcome including expansion of the CAR T-cell culture (fold expansion), expansion of the CAR T cells after infusion (qPCR CAR days 0-35), persistence of CAR T cells calculated at days 0 to 35 postinfusion (AUC days 0-35), and overall expansion of CD8+ T cells after infusion (CD3+CD8+ peak days 0-28) (CR, n = 6; NR, n = 18). (C) Mitochondrial mass of CAR+CD8+ T cells plotted against the percentage of CD27+ cells negative for PD-1, TIM-3, and LAG-3 (CR, n = 6; NR, n = 18). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Mann-Whitney test (A). Correlations were determined by a Spearman Rho test (B-C). Data are presented as mean plus or minus SEM. *P < .05.
Discussion
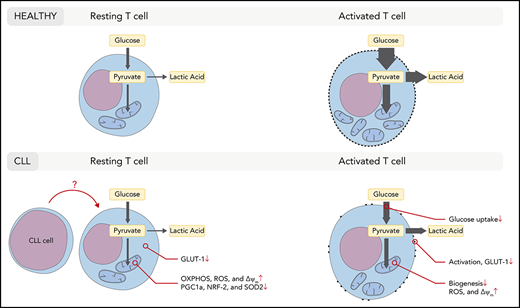
T cells in CLL acquire a dysfunctional state as CLL progresses through as-yet not fully elucidated mechanisms.57 We and others have previously shown that T-cell metabolism is tightly linked to T-cell differentiation and function.27-31,47 Furthermore, we have demonstrated that CD8+ T cells in CLL exhibit reduced glycolytic activity after stimulation.38 Here, we focused on the metabolic aspects of resting CD8+ T cells that might impact on the metabolic reprogramming upon stimulation, and found that CD8+ T cells in CLL have lower GLUT1 reserves and exhibit a skewed mitochondrial metabolic profile prior to stimulation, which is maintained after stimulation.
Previous work has shown that tumor cells can compete in a metabolic “tug of war” with tumor-infiltrating lymphocytes (TILs) for nutrients, which impedes glucose uptake by T cells.34 Although we observed impaired glucose uptake in CD8+ T cells when exposed to CLL cells, we found that the impaired glycolytic response upon stimulation could not be attributed to competition for glucose, nor to suppressive effects of lactate. This is supported by the fact that surface GLUT1 expression is restored when CLL cells were depleted. These findings indicate that, in contrast to many other tumor cells, CLL cells themselves are not highly glycolytic. It has indeed been described that circulating CLL cells display similar glycolytic activity compared with healthy B cells,58 which is also supported by the generally low maximum standardized uptake values as measured by fluorodeoxyglucose positron emission tomography/computed tomography scanning in CLL patients.16 Despite the absence of glucose competition between CD8+ T cells and CLL cells, CLL cells were able to reduce glycolysis in CD8+ T cells while physically separated but present in the same culture, suggesting that inhibition of glycolysis may be explained by a soluble factor. Immunosuppressive cytokines such as interleukin 10 (IL-10) and transforming growth factor β have been described to be produced by CLL cells,59-64 and it was recently reported that transforming growth factor β represses several metabolic pathways in natural killer cells.65 Although a direct link between IL-10 and metabolic plasticity in T cells has not yet been described, this cytokine opposes the switch to the metabolic program induced by inflammatory stimuli in macrophages.66 Interestingly, production of IL-10 during prolonged treatment with ibrutinib is reduced, which correlated with improved T-cell numbers and function.67
Upon T-cell activation, increased amounts of glucose must be transported into the cell via glucose transporters, of which GLUT1 is the most abundant.42 Although in resting CD8+ T cells expression of surface GLUT1 and glucose uptake was comparable between CLL patients and HDs, intracellular vesicular storages of GLUT1 are reduced in CLL. This observation may explain why CLL CD8+ T cells fail to upregulate surface GLUT1 upon stimulation, and therefore have impaired glucose uptake. The lower GLUT1 reserves could not be explained by reduced GLUT1 RNA or HIF-1α levels. In contrast, we found higher HIF-1α expression in CLL-derived CD8+ T cells, which is probably a result of the higher mitochondrial ROS levels in those cells because ROS stabilizes HIF-1α.46 These data hint toward posttranscriptional processes being involved in reduced vesicular GLUT1 expression in CLL-derived CD8+ T cells.46,68 Our previous study indeed demonstrated reduced total GLUT1 protein after stimulation.38 This further implies that a GLUT1 production problem rather than a trafficking problem causes the reduced surface expression of GLUT1. Nevertheless, impaired vesicular trafficking has been described in CLL T cells,25 and whether GLUT1 trafficking to the cell surface is affected remains to be determined.
We observed that resting CD8+ T cells in CLL exhibit a skewed mitochondrial metabolic profile as demonstrated by elevated OXPHOS, but decreased SRC compared with HDs. Our data further demonstrate that CD8+ T cells in CLL have high ΔΨm and produce greater amounts of mitochondrial ROS compared with HDs, analyzed directly ex vivo and after activation. T cells with high ΔΨm have been found to be associated with a terminally differentiated state as these cells exhibit low expansion and poor tumor-killing capacity.49 In contrast, T cells with low ΔΨm exhibit a memory phenotype, show robust expansion, and effectively mediate tumor cell killing.49 Although mitochondrial ROS is essential for T-cell activation,39 excessive production of ROS by mitochondria inhibits T-cell development and function.69 Similar to our observations in CLL-derived T cells, human clear cell renal cell carcinoma CD8+ TILs were unable to efficiently take up glucose, and generated large amounts of mitochondrial ROS, which was associated with downregulated SOD2. Improved activation of these clear cell renal cell carcinoma TILs was observed by reducing mitochondrial ROS using mitochondrial antioxidants.70 Similarly, suppressing mitochondrial ROS could also restore antiviral activity of exhausted hepatitis B virus-specific CD8+ T cells in chronic hepatitis B, further demonstrating the importance of improving mitochondrial health to boost T-cell function.71 These observations and the data we present in this study suggest that deteriorated mitochondrial health may be the basis of global dysfunction in T cells in CLL. In many cell types, PGC-1α is the master regulator of mitochondrial function and biogenesis.72,73 We found reduced expression of PGC-1α in resting CD8+ T cells in CLL. In a mouse model of melanoma, overexpression of PGC-1α increased mitochondrial mass in CD8+ T cells and improved tumor-killing capacity.50 In line with this, mitochondrial mass was reduced in CLL-derived antigen-experienced CD8+ T cells analyzed directly ex vivo, and mitochondrial biogenesis was impaired in CLL-derived CD8+ T cells upon stimulation in vitro.
Clinical responses of CD19-CAR T-cell therapy in CLL can be dramatic but only occur in a minority of patients.11,13-15 Here, we demonstrate that mitochondrial mass was higher in CRs, and correlated with expansion of CAR T cells in culture and their persistence in vivo. Furthermore, we found that mitochondrial mass correlated with the presence of CD27+ T cells that were negative for PD-1, TIM-3, and LAG-3. Similar to our results, it has previously been described that loss of mitochondrial mass correlates with upregulation of co-inhibitory markers PD-1, TIM-3, and LAG-3.50 Our data showing impaired mitochondrial biogenesis in CAR T cells from NR patients are in line with our results on CLL-derived CD8+ T cells showing reduced mitochondrial mass in steady state and impaired mitochondrial biogenesis upon stimulation. However, we did not observe any differences in ΔΨm, mitochondrial ROS, and glucose uptake between CR and NR patients. Differences in observations between the CAR T cells vs CLL-derived CD8+ T cells can most likely be explained by differences in culturing methods (9-11 days vs 2 days), cell type composition (absence of CLL cells vs ±90% CLL cells), and treatment status (heavily treated vs untreated patients). The CAR described in the current studies is a second-generation CAR containing the 4-1BB domain as a costimulatory domain. This costimulatory molecule was recently described to induce mitochondrial biogenesis, promote T-cell central memory differentiation, and to improve immunotherapeutic approaches including CAR T-cell therapy.74-76 Moreover, CD19 CAR+CD8+ T cells generated in memory stem cell–enriched conditions possessed more SRC and provided long-lasting antitumor responses.77 We therefore postulate that strategies that promote mitochondrial biogenesis during CAR T-cell generation might ultimately benefit the efficacy of this therapy in CLL and possibly other B-cell malignancies. Because the process of CAR T-cell generation involves transduction of a CAR-encoding construct and has an vitro expansion phase, these strategies can include both genetic and pharmacologic interventions on key metabolic players such as the nicotinamide adenine dinucleotide–sirtuin 1–5′ AMP-activated protein kinase–PGC-1α axis.78-80
Presented during the 60th annual meeting of the American Society of Hematology, San Diego, CA, 1-4 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
For original data, please contact the corresponding authors. Microarray data are available at GEO under accession number GSE8835.
Acknowledgments
The authors thank the patients and healthy donors for their blood donations. The authors also thank Bart Everts for carefully reading the manuscript.
This work was supported by a Netherlands Organisation for Scientific Research (NWO)/Netherlands Organisation for Health Research and Development (ZonMw) VENI grant and a European Union Marie Curie reintegration grant (G.J.W.v.d.W.), an NWO/ZonMw VIDI grant (A.P.K.), an Amsterdam Infection and Immunity Institute (AI&II) travel grant (J.A.C.v.B.), National Institutes of Health National Cancer Institute grant RO1CA226983 (C.H.J.), and the Else Kröner Fresenius Stiftung (P.J.S.).
Authorship
Contribution: J.A.C.v.B., J.A.F., S.H.T., E.E., P.J.S., J.C.R., J.J.M., A.P.K., and G.J.W.v.d.W. designed research; J.A.C.v.B., A.W.J.M., S.E., T.H., and G.J.W.v.d.W. performed research; J.A.C.v.B. and G.J.W.v.d.W. analyzed data; J.A.F., M.-D.L., D.L.P., J.J.M., and C.H.J. provided patient samples and reviewed the paper; and J.A.C.v.B., A.P.K., and G.J.W.v.d.W. wrote the paper.
Conflict-of-interest disclosure: J.A.F., D.L.P., J.J.M., and C.H.J. have sponsored research grants from Novartis and are inventors on patents related to CAR T-cell therapy. The remaining authors declare no competing financial interests.
Correspondence: Arnon P. Kater, Department of Hematology, Amsterdam UMC, University of Amsterdam, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: a.p.kater@amc.nl; and Gerritje J. W. van der Windt, Genmab, Uppsalalaan 15, 3584 CT Utrecht, The Netherlands; e-mail: riw@genmab.com.

![Figure 1. Impaired activation and reduced glucose uptake by CLL-derived CD8+ T cells upon stimulation. PBMCs from CLL patients and age-matched HDs were thawed and cultured for 2 (A-B,D-F) or 5 (C) days in the presence or absence of anti-CD3/CD28 antibodies. Cells were analyzed for (A) activation markers (CLL, n = 15-19; HD, n = 16-22), (B) degranulation (CLL, n = 12; HD, n = 12), (C) PD-1 expression (CLL, n = 8; HD, n = 8), (D) surface glucose transporter GLUT1 (measured by GLUT1 Ras-binding domain [RBD] green fluorescent protein [GFP] construct) (CLL, n = 6; HD, n = 4; data are the same as in supplemental Figure 1G), and (E) glucose uptake (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose [2-NBDG]; CLL, n = 23; HD, n = 20; data are the same as in supplemental Figure 1F), and expression of (F) HIF-1α (CLL, n = 6; HD, n = 4). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by an unpaired Student t test (A-C,E), or a Mann-Whitney test (A,D,F). Data are presented as mean plus or minus standard error of the mean (SEM). *P < .05; **P < .005; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/1/10.1182_blood.2018885863/3/m_blood885863f1.png?Expires=1770145993&Signature=tvNkZWYnjQI1FR~fUj81o-S0u5tGNdBfzfDcIYdHAqM2TAnq2XGPJgJj~9eZT8u3eqB4gjNplX4oaFnjdZrNIHeCW~AYHABJ6wMu9nKOnLoerg7~686aj2yJ27JRareuyuB2uEOoriWrgQb8Uzo~jyADhrAbHtH2oO~5KAzCGE2p6SQgrJq8P62ExF0hK1KS3Zk6EhPvhrr5sv~gBrWjshoz3zd6cBQZHtJVbjHTpmOGaHROpcKJEVB7Ga41GGiAwb7vimDXXdIeDi8S3cILtuiuads-x4j75ZDAiZm-pOwQjvIm9k3bpBORsujXAEonN-YsFRV39F1bdOo2HYwp3w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Figure 4. Altered mitochondrial homeostasis in CLL-derived CD8+ T cells. (A) KEGG pathway analysis showing top 10 of most significantly different pathways in CD8+ T cells from HDs vs CLL, and heatmap of differentially expressed genes in OXPHOS pathway (Gene Expression Omnibus [GEO] accession number GSE8835; P = 3.4 × 10−15 for OXPHOS pathway). PBMCs from CLL patients and age-matched HDs were analyzed by flow cytometry (C,E-H) or Seahorse EFA (B,D) either directly after thawing (B-D,H), 2 days of culturing (E-F), or 3 days of culturing (G). (B) Cells were analyzed for OCR (indicating OXPHOS; CLL, n = 7; HD, n = 6). (C) Mitochondrial mass was determined by flow cytometry (Mitotracker Green; CLL, n = 29; HD, n = 29) and by qPCR by calculating the mitochondrial DNA (mtDNA)-to-nuclear DNA (nDNA) ratio (CLL, n = 8; HD, n = 5). (D) SRC was calculated as the ratio of maximum OCR over basal OCR (CLL, n = 6; HD, n = 6). (E) ΔΨm was determined by flow cytometry (Mitotracker Orange; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5D). (F) Mitochondrial ROS (mitoSOX; CLL, n = 11; HD, n = 12; data are the same as unstimulated control in Figure 5E). (G) Mitochondrial ROS as well as mitochondrial potential were analyzed in HD CD8 T cells cocultured with HD-derived B cells or CLL (HD, n = 8). (H) PGC-1α, SOD1, and SOD2 (CLL, n = 8; HD, n = 8), HO-1, NRF-2, and ERRα (CLL, n = 8; HD, n = 4). Data were normalized to HD to compile multiple independent experiments (B,D). Normality was determined by a D’Agostino and Pearson normality test. The P value was calculated by a Welch test (E-F), Mann-Whitney test (B-D,G-H), or an unpaired Student t test (H). Data are presented as mean plus or minus SEM. *P < .05; **P < .005; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/1/10.1182_blood.2018885863/3/m_blood885863f4.png?Expires=1770145993&Signature=aK5jm8g4M7ba6yC~70VU~b5YCkyYAeEvkhlv2eH2pqFZZxSQ4BRpONI2YAjzk4LObBRT2CnbimxfooY1qhyzv5bmibIGwNsZ9nZTyHAJIZNFGqyxtVfj2GWTGysDutRafU~5W~V51KP0PP8IxqpViKIRWtLsXsUZH7C5B59QwaOndolWUp1Y4Ce28LUklGN4Ls9cCCgK482w8UqD0GbuKlCQDosTBUcw1K~-13RcUYQSHKHmFOcTi4liQtTGFLT5RM6uOzY-44G3cejzccNwdsaqOL3c4KHJx85bpD3wYGwBk9FJTSpeCDObXtu27YdUb6wN8nfJggFt2kHQw1-OCA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)