

TO THE EDITOR:
Natural killer (NK)–cell enteropathy, also referred to as lymphomatoid gastropathy, is a recently described, rare indolent NK-cell lymphoproliferative disorder (LPD) that involves single or multiple sites along the gastrointestinal tract.1,-3 Patients often present with vague gastrointestinal symptoms, including abdominal pain, constipation, diverticulosis, and reflux, but they have no prior history of celiac disease, inflammatory bowel disease, or malabsorption. The lesions exhibit a chronic, relapsing clinical course, and usually do not show prolonged response to chemotherapy. A distinction from aggressive NK or T-cell lymphoma is of paramount importance to avoid unnecessary aggressive therapies. The pathogenesis of NK-cell enteropathy remains unknown, and whether it represents a true neoplastic process is still unresolved, partly because of the challenges of demonstrating clonality in NK cells. In this study, we characterize the clinicopathologic features and investigate the mutational profiles and related aberrant signal pathways in this rare entity.
Among the 10 patients, 1 (case 3) was included in our initial series,3 and the other 9 patients were newly diagnosed (Table 1). There were 7 females and 3 males with a median age of 61 years (range, 9-76 years). This research was approved by the Memorial Sloan Kettering institutional review board. The initial symptoms included abdominal pain/discomfort,4 reflux,1 and diarrhea.1 Three patients were diagnosed during screening colonoscopy examination. The sites of involvement were stomach,5 duodenum,6 colon,4 and small intestine (3, including 2 terminal ileum). Endoscopic findings included superficial ulcers and/or erosions in 5/7 patients and polyps in 2/7 patients. The only pediatric patient (case 8) had a mildly enlarged cervical lymph node that was deemed unrelated. All other patients had no lymphadenopathy or hepatosplenomegaly. One patient (case 10) had positron emission tomography-computed tomography imaging showing an standardized uptake value of 4 at the gastric fundus. The key histologic features were expansion of the lamina propria by a relatively well-circumscribed but confluent infiltrate of medium-sized cells with irregular nuclei, inconspicuous nucleoli, finely clumped chromatin, and a moderate amount of pale cytoplasm (Figure 1A-B). The mucosal glands were displaced in 2 patients (cases 1 and 3) because of the dense atypical cellular infiltrate, but glandular destruction was not seen. Increased intraepithelial lymphocytes were only seen in 1 patient (case 8). Angiocentricity or angiodestructive pattern of growth was absent. Necrosis was not seen. The muscularis mucosae were intact. There was no villous atrophy or crypt hyperplasia. Other inflammatory cells such as plasma cells, neutrophils, and eosinophils were often present. Immunophenotypically, the cells were positive for CD56 (10/10), CD2 (5/8), cytoplasmic CD3 (9/9), TIA-1 (10/10), granzyme B (10/10), and CD7 (5/6; Table 1; Figure 1C-E; data not shown). Partial CD8 expression was seen in 1 case (case 10). The cells were negative for CD4, surface CD3, CD5, T-cell receptor β (TCRβ), and TCRγ or TCRδ (data not shown). Epstein-Barr virus–encoded small RNA (EBER) in situ hybridization was negative (10/10; Figure 1F). Helicobacter pylori was positive by serology in 1 case (case 1), negative by immunostains in 2 cases (cases 2 and 10), and negative in all 10 cases by morphology. Ki-67 proliferation index was examined in 8 patients and was variable: high (75%-90%) in 2 cases, moderate (50%-75%) in 1 case, and intermediate (25%-50%) in 5 cases. Clonal gene rearrangements of both TCRβ and TCRγ were initially reported to be positive in 1 patient (case 10), and the patient was misdiagnosed as having aggressive NK/T-cell lymphoma and was prepared for chemotherapy and hematopoietic stem cell transplantation. Repeated PCR studies of the same biopsy and an additional biopsy specimen confirmed a polyclonal pattern. Therefore, TCRγ gene rearrangements were polyclonal or showed a restricted pattern in 10/10 patients. A second patient (case 6) was also misdiagnosed as having T-cell lymphoma, based on the observed expression of CD3, and received a cycle of chemotherapy. Follow-up data were available in 8 patients. Median follow-up was 59.5 months. Repeat biopsy was performed in 4 patients and showed persistent disease with a similar anatomic distribution and pathological features. None of the patients had progression beyond the gastrointestinal tract or died of disease.
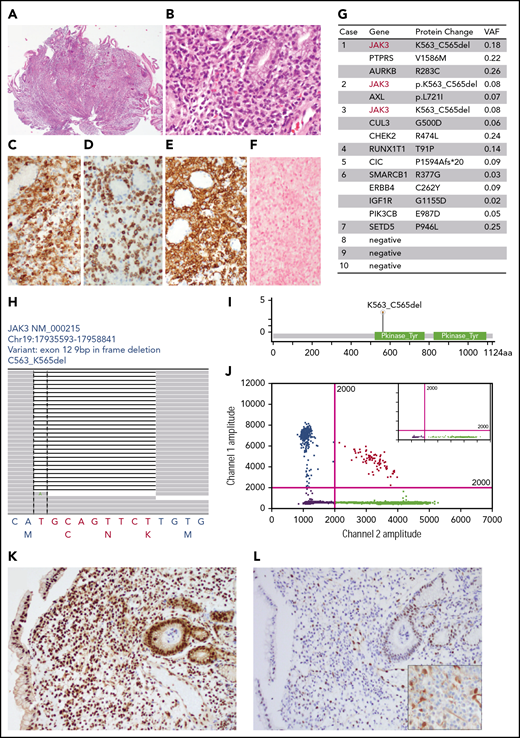
The histologic and mutational profiles of NK-cell enteropathy (all data from case 2 except panel J from case 3). (A-B) Hematoxylin and eosin histologic pictures. (C-E) Immunohistochemical stains of CD2 (C), cytoplasmic CD3 (D), and CD56 (E). (F) EBER in situ hybridization. (G) Somatic mutations identified by targeted next-generation sequencing panel (MSK heme IMPACT). (H) Snapshot of the Integrative Genomic Viewer window showing an in-frame deletion (K563_C565) in exon 12 of JAK3. The bottom letters show bases and amino acids. (I) Lollipop illustration of JAK3 K563_C565del mutation. (J) Representative graph of droplet digital PCR studies. x-axis, WT allele; y-axis, JAK3 K563_C565del allele. Inset, normal control. (K-L) Immunohistochemical results of anti-p-STAT5 (K), anti-p-STAT3 (L), and anti-p-ERK (L inset) in NK-cell enteropathy.
The histologic and mutational profiles of NK-cell enteropathy (all data from case 2 except panel J from case 3). (A-B) Hematoxylin and eosin histologic pictures. (C-E) Immunohistochemical stains of CD2 (C), cytoplasmic CD3 (D), and CD56 (E). (F) EBER in situ hybridization. (G) Somatic mutations identified by targeted next-generation sequencing panel (MSK heme IMPACT). (H) Snapshot of the Integrative Genomic Viewer window showing an in-frame deletion (K563_C565) in exon 12 of JAK3. The bottom letters show bases and amino acids. (I) Lollipop illustration of JAK3 K563_C565del mutation. (J) Representative graph of droplet digital PCR studies. x-axis, WT allele; y-axis, JAK3 K563_C565del allele. Inset, normal control. (K-L) Immunohistochemical results of anti-p-STAT5 (K), anti-p-STAT3 (L), and anti-p-ERK (L inset) in NK-cell enteropathy.
To examine the mutational profile, DNA was extracted from formalin fixed paraffin embedded biopsy tissue and submitted for sequencing studies, using a targeted next-generation sequencing panel covering 576 genes known to be mutated in hematologic malignancies (MSK Heme IMPACT),4 Two patients (cases 2 and 10) had germline controls (blood and nails). The sequencing results were evaluated by a custom pipeline, and somatic mutations were called on the basis of a pooled normal or patient-specific normal controls. Somatic mutations were identified in 7/10 cases (Figure 1G; supplemental Table 1, available on the Blood Web site). The variant allele frequencies ranged from 2% to 25%. Cases 3 and 6 also showed subclonal mutations. Therefore, the tumor content was estimated as 15% to 50% by allelic frequencies, which was confirmed by immunohistochemical findings. The most important finding was detection of identical JAK3 mutations (K563_C565del) in 3/10 patients (Figure 1H-I). Other mutations identified in this cohort are shown in Figure 1G and supplemental Table 1. These included PTPRS, AURKB, AXL, ERBB4, IGF1R, PIK3CB, CUL3, CHEK2, RUNX1T1, CIC, SMARCB1, and SETD5. Data are available on National Center for Biotechnology Information ClinVar, under accession numbers ClinVar SCV000930594-ClinVar SCV000930606.
Since JAK3 K563_C565del mutations were present in 30%, we designed a droplet digital PCR primer set specific for this mutation to confirm the results. As shown in Figure 1J, wild-type (WT) control sample only gave rise to WT signals, whereas both WT and JAK3 K563_C565del alleles were detected in 2 patients (case 1 and 3) in whom DNA was available for analysis. This mutation localizes to the pseudokinase domain that normally provides inhibitory feedback to suppress JAK kinase activity. Therefore, mutations in this domain nearly universally lead to upregulation of JAK kinase activation. Indeed, this JAK3 K563_C565del mutation induces STAT5 phosphorylation in T-prolymphocytic leukemia.6,7 Because of the limited material available for study, we performed immunohistochemical studies to examine phospho-molecules including p-ERK, p-STAT3, and p-STAT5. Intense signals were detected using anti-p-STAT5 antibody indicating an activation of STAT5 in 7/7 patients (Figure 1K). Only 2 cases had tissue for additional immunohistochemical studies of p-ERK and p-STAT3: 1 had JAK3 K563_C565del mutation (case 2), and the other had no mutations detected (case 10). Signals of p-ERK and p-STAT3 were not detected in neoplastic cells in either case (Figure 1L).
In summary, we report the mutational profile of NK-cell enteropathy for the first time. We demonstrate recurrent JAK3 K563_C565del mutations in 30% (3/10) of cases. Multiple studies have shown that mutations in the JAK-STAT pathway occur in up to 30% of NK or T-cell lymphomas.7,,,,-12 JAK3 K563_C565del mutations have been reported in T-LPD/lymphoma, particularly T-PLL.6,7 Identification of the same mutation in a wide variety of diseases ranging from indolent to aggressive suggests that additional molecular mechanisms may contribute to these diseases. These mutations lead to activation of downstream targets such as STAT3 and STAT5.5,7 Activation of p-STAT5 was seen in all tested cases in our study, including the ones without JAK3 K563_C565del mutation, suggesting that alternative mechanisms other than JAK3 mutation may activate STAT5. In this regard, the mutations in genes that regulate tyrosine kinase-based signaling pathways might lead to STAT5 activation such as PTPRS, AURKB, AXL, ERBB4, IGF1R, and PIK3CB. Mutations in PTPR, CHEK2, SMARCB1, ERBB4, PIK3CB, and SETD5 have also been reported in T-cell lymphoma/leukemia.9,13,14 CHEK2, SMARCB1, and SETD5 regulate DNA damage responses. AURKB encodes aurora kinase B that regulates mitosis and DNA damage responses. CUL3 encodes cullin 3, part of ubiquitin ligase complex. AXL is a TAM family member that is expressed on NK cells and plays important role in NK-cell development.15
The identification of recurrent mutations in NK-cell enteropathy support a neoplastic origin, and it may be considered a distinct neoplastic disease entity with the following diagnostic criteria: indolent clinical behavior but chronic, recurring course; dense superficial mucosal lymphoid infiltrate by lymphoid cells with mild cytological atypia; NK immunophenotype; lack of TCR gene arrangements and EBV. Although the clinical presentations are largely overlapping, NK-cell enteropathy needs to be differentiated from indolent T-cell LPD of the gastrointestinal (GI) tract.16,17 Progression to more aggressive disease has been reported in some cases of indolent T-cell LPD of the GI tract, but not in NK-cell enteropathy. Both share a similar morphologic pattern of superficial mucosal infiltrate, but an NK immunophenotype and lack of TCR gene rearrangements are essential for a diagnosis of NK-cell enteropathy. In addition, NK-cell enteropathy may show superficial ulceration.
Although NK-cell enteropathy has an indolent clinical course, the lesions in general do not respond to treatment and tend to be persistent or recur, and the patients can be symptomatic. The newly identified JAK3 mutations and activation of STAT5 pathway may represent a potential therapeutic target, particularly for patients with persistent symptoms.
Part of the data were presented in abstract form at the 19th meeting of the European Association for Haematopathology, Edinburgh, Scotland, 29 September-4 October 2018.
The online version of this article contains a data supplement.
Acknowledgments
This study was supported in part through the National Institutes of Health, National Cancer Institute Cancer Center Support Grant P30 CA008748 and intramural grant from the National Institutes of Health, National Cancer Institute Cancer. W.X. is supported by a startup fund from Department of Pathology at Memorial Sloan Kettering Cancer Center.
Authorship
Contribution: W.X., G.K.G., and E.S.J. conceived the study, collected and analyzed the data, and wrote the manuscript; J.B., A.S., and S.P. collected data; J.Y., Y.J.J., L.X., M.E.A., and M.R. performed the sequencing studies and annotated the sequencing data; A.K. and A.J.M. provided critical clinical information; A.D. interpreted the data; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: A.D. has received personal fees from Roche, Corvus Pharmaceuticals, Physicians' Education Resource, Seattle Genetics, Peerview Institute, Oncology Specialty Group, Pharmacyclics, Celgene, and Novartis and research grants from the National Cancer Institute and Roche. The remaining authors declare no competing financial interests.
Correspondence: Wenbin Xiao, Hematopathology Diagnostic Service, Department of Pathology, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: xiaow@mskcc.org; and Elaine S. Jaffe, Section of Hematopathology, Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892; e-mail: ejaffe@mail.nih.gov.

