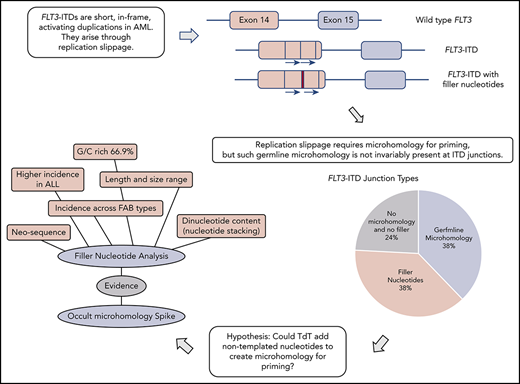
Key Points
FLT3-ITDs unexpectedly show junctional N-nucleotides with properties consistent with synthesis by TdT.
Off-target TdT activity in AML is proposed to promote FLT3-ITD formation by priming replication slippage.

Abstract
FLT3–internal tandem duplications (FLT3-ITDs) are prognostic driver mutations found in acute myeloid leukemia (AML). Although these short duplications occur in 25% of AML patients, little is known about the molecular mechanism underlying their formation. Understanding the origin of FLT3-ITDs would advance our understanding of the genesis of AML. We analyzed the sequence and molecular anatomy of 300 FLT3-ITDs to address this issue, including 114 ITDs with additional nucleotides of unknown origin located between the 2 copies of the repeat. We observed anatomy consistent with replication slippage, but could only identify the germline microhomology (1-6 bp) anticipated to prime such slippage in one-third of FLT3-ITDs. We explain the paradox of the “missing” microhomology in the majority of FLT3-ITDs through occult microhomology: specifically, by priming through use of nontemplated nucleotides (N-nucleotides) added by terminal deoxynucleotidyl transferase (TdT). We suggest that TdT-mediated nucleotide addition in excess of that required for priming creates N-regions at the duplication junctions, explaining the additional nucleotides observed at this position. FLT3-ITD N-regions have a G/C content (66.9%), dinucleotide composition (P < .001), and length characteristics consistent with synthesis by TdT. AML types with high TdT show an increased incidence of FLT3-ITDs (M0; P = .0017). These results point to an unexpected role for the lymphoid enzyme TdT in priming FLT3-ITDs. Although the physiological role of TdT is to increase antigenic diversity through N-nucleotide addition during V(D)J recombination of IG/TCR genes, here we propose that illegitimate TdT activity makes a significant contribution to the genesis of AML.
Introduction
FLT3 encodes a receptor tyrosine kinase that governs proliferation of early progenitor hematopoietic cells.1 FLT3–internal tandem duplications (FLT3-ITDs) are 1 of the commonest mutations found in acute myeloid leukemia (AML),1,2 and also occur occasionally in acute lymphoblastic leukemia (ALL).3,4 In AML, they confer a poor prognosis, particularly at higher allelic loads5,6 arising from acquired isodisomy,7 although development of effective FLT3 inhibitors may improve this outlook. FLT3-ITDs vary in size from 3 to over 200 bp, invariably remain in-frame,6 and target exon 14, resulting in constitutive kinase activation. Both start and end points are variable, with the majority of duplications considered unique.8 Patients have been reported with up to 5 independent ITDs,9-11 consistent with a mutator mechanism that promotes their formation.
Genomic rearrangements can be characterized through examination of junction sequences for recombinogenic motifs and homology.12-15 Despite the importance of FLT3-ITDs in AML, the mechanism underlying their formation has not been addressed beyond proposition of a replication error.16 Replication-based models (microhomology-mediated replication dependent recombination [MMRDR]) encompass both simple replication slippage, typically involving change of a few bases at a repetitive sequence, and more complex models requiring breakage of the template strand.12,17 If a replication model of FLT3-ITD genesis is to be accepted, then the microhomology required for priming slippage should be identified. Moreover, any proposed model must also account for the frequent insertion of nucleotides of unknown origin at the point of duplication, referred to as filler or foreign DNA.2,6,18
Failure to identify germline microhomology might argue against a replication-based origin for FLT3-ITDs. However, a suggestion for how microhomology can be created comes from the double-strand break repair process termed nonhomologous end joining (NHEJ). Microhomology is not essential for NHEJ to join ends, but enhances ligation if present. In rare instances, this microhomology can be provided through nontemplated nucleotides (N-nucleotides) added by polymerase X family members (Pol μ, Pol λ, or terminal deoxynucleotidyl transferase [TdT]).19,20 As this homology is not present in the original sequences it usually escapes detection, and is therefore known as occult microhomology. However, occult microhomology remains poorly understood, with no identified role in MMRDR.
Here, we investigate the origin of FLT3-ITDs through examination of their molecular anatomy and breakpoint sequences. We propose a model of replication slippage primed extensively by occult microhomology synthesized by TdT, with a supporting role for germline microhomology. Longer TdT additions are revealed as N-regions at the duplication junction. We propose that the purposefully mutagenic enzyme TdT plays a significant role in the genesis of AML.
Methods
Patient cohort
Genomic DNA or complementary DNA FLT3-ITD sequences were identified using PubMed and Google publications from 1996 to 2013 (supplemental Table 1, available on the Blood Web site). Exclusion criteria were uncertain reference sequence, suspected contamination, and repeat publication. Numbering was converted to the FLT3 coding reference sequence LRG_457t1. Breakpoint positions were harmonized to the Human Genome Variation Society (HGVS) 3′ rule, and to recognize triplications or intronic end points. Three hundred ITDs (AML, n = 273; chronic myelomonocytic leukemia, n = 6; myelodysplastic syndrome, n = 4; acute mixed lineage leukemia, n = 1; ALL, n = 16) were identified from 275 patients, representing 271 primary samples and 4 cell lines. For 19 of 114 ITDs with filler, the ITD position and filler length were available but the sequence of the filler was not stated. These ITDs were retained to preserve the proportion of patients with fillers. The sequence of 7 of 95 of the remaining fillers was partially deduced by reverse translation. Five additional FLT3-ITDs from ALL were considered only with respect to filler nucleotide incidence. French-American-British (FAB) type was available for 220 of 273 AML FLT3-ITDs.
Statistics and homology searches
Cumulative binomial probabilities and Fisher exact test 2-sided P values were calculated at www.danielsoper.com/statcalc. The Spearman rank correlation was performed at www.wessa.net/rwasp_spearman.wasp/. P values of <.05 were considered significant, unless adjusted by Bonferroni correction. Basic Local Alignment Search Tool (BLAST) searches were performed at National Center for Biotechnology Information (NCBI; http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Results
Molecular anatomy and incidence of FLT3-ITDs
The 300 FLT3-ITD sequences were divided according to molecular anatomy (Figure 1). Types A-C were duplications, each starting within exon 14, and ending within exon 14, intron 14, and exon 15, respectively (length range, 18-240 bp; mean, 57 bp). Type D showed insertion of filler sequence (length range, 3-18 bp; mean, 10 bp), without loss or gain of FLT3 material, suggesting there is no critical region of FLT3 that must be duplicated. Type E were deletions, although 3 of 12 appeared larger than wild type due to insertion of filler that exceeded the deleted material in length. A single-type E mutation was length neutral, with deletion of FLT3 sequence balanced in length by gain of filler. Types F and G represented more complex events, including full or partial triplications. MMRDR could account for all types via backward slippage (duplication), forward slippage (deletion), and repeat slippage (more complex events including triplications). We therefore looked for the obligatory junction microhomology required to substantiate a replication-based model.
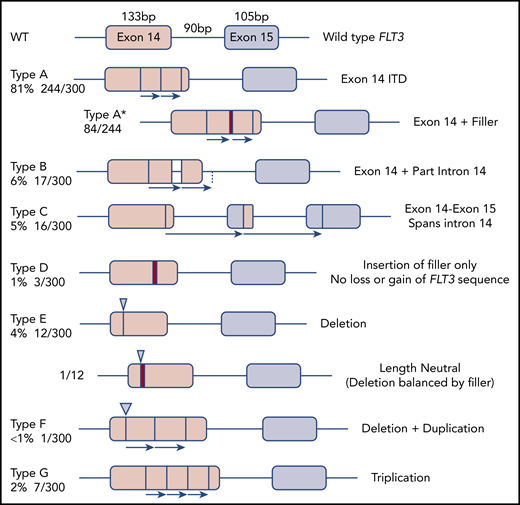
Molecular anatomy and incidence of different types of FLT3-ITD. Only types A, B, and C meet the HGVS definition of a duplication, unless a filler (N-region, indicated by a red bar in the diagram and an asterisk (*) in the nomenclature) is present at the junction. All types can have N-nucleotides, type D by definition.
Molecular anatomy and incidence of different types of FLT3-ITD. Only types A, B, and C meet the HGVS definition of a duplication, unless a filler (N-region, indicated by a red bar in the diagram and an asterisk (*) in the nomenclature) is present at the junction. All types can have N-nucleotides, type D by definition.
Identification of 3 junction categories
Breakpoint examination did not identify universal flanking microhomology. Instead, 3 different junction categories were identified: 38% of ITDs showed the anticipated germline microhomology (1-6 bp), 38% showed addition of nontemplated filler nucleotides of unknown origin, and 24% lacked either microhomology or filler nucleotides (supplemental Table 2). Each junction type was identified across the mutational spectrum (duplications, deletions, and triplications), arguing against separate mechanistic origins for the different ITD types. All 3 junction types were identified in both myeloid- and lymphoid-derived ITDs (supplemental Table 2). The choice of junction type could vary between multiple independent ITDs identified from a single patient; 1 patient showed examples of each category. Junction category was not necessarily conserved at both junctions found in triplications. Each junction category is explored in the following 3 sections.
Germline microhomology–mediated junctions
Figure 2A shows how microhomology can prime MMRDR, consistent with the 1 to 6 bp of microhomology observed flanking 38% of junctions (supplemental Tables 2 and 3). Although some microhomology would be expected by chance, there is an excess of microhomology at each length over that expected by chance (supplemental Figure 1). The observed excess increases with increasing microhomology length.
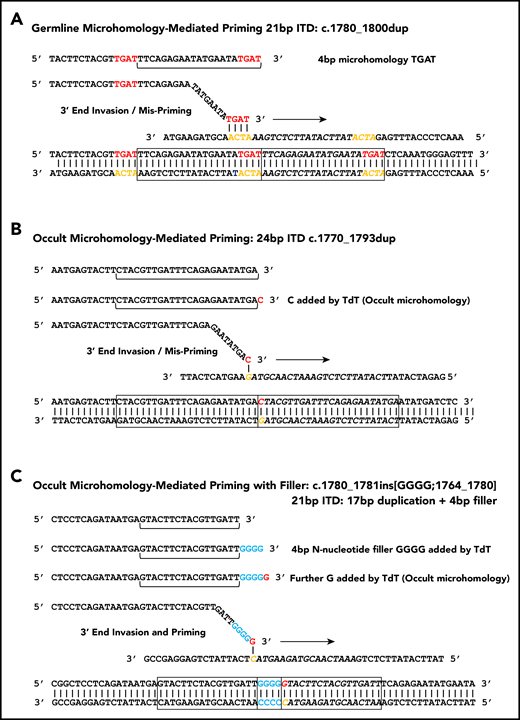
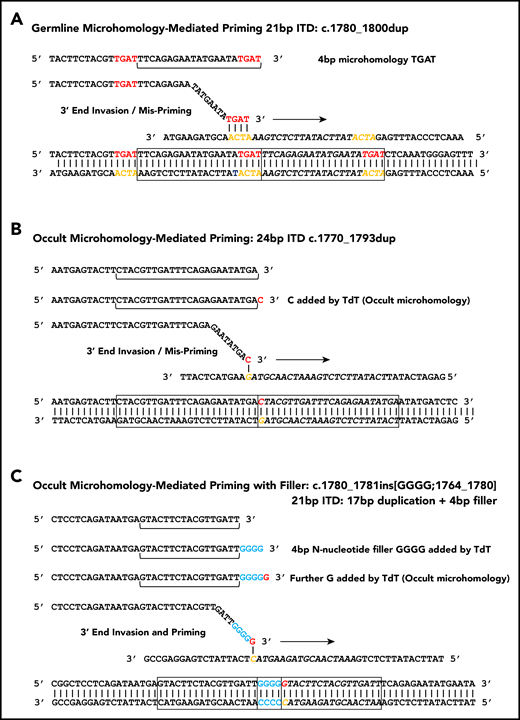
Microhomology for priming FLT3-ITD replication slippage. (A) Misalignment and priming using germline (visible) microhomology. The recurrent c.1780-1800dup primed by TGAT microhomology (red against yellow) is shown. (B) Occult (invisible) microhomology-mediated priming, where TdT adds a base to the misaligning 3′ end. This base provides the homology for priming, but is not detected in the final sequence as it matches an existing base. The 24-bp c.1770-1793dup is shown. (C) Occult microhomology-mediated priming following addition of multiple N-nucleotides (blue). Only the terminal N-nucleotide matches the target strand, hence the previous N-nucleotides are visible as filler DNA between the 2 copies of the repeat. A 21-bp FLT3-ITD is shown (17-bp c.1764_1780 duplication, with 4 additional N-nucleotides). These models are applicable to either simple replication slippage or break-induced models of MMRDR. The latter can occur following replication across a single-stranded nick and resulting replication fork collapse; fork collapse creates a single-ended double-stranded break, and the 3′ end must invade the template strand to reinitiate replication.
Microhomology for priming FLT3-ITD replication slippage. (A) Misalignment and priming using germline (visible) microhomology. The recurrent c.1780-1800dup primed by TGAT microhomology (red against yellow) is shown. (B) Occult (invisible) microhomology-mediated priming, where TdT adds a base to the misaligning 3′ end. This base provides the homology for priming, but is not detected in the final sequence as it matches an existing base. The 24-bp c.1770-1793dup is shown. (C) Occult microhomology-mediated priming following addition of multiple N-nucleotides (blue). Only the terminal N-nucleotide matches the target strand, hence the previous N-nucleotides are visible as filler DNA between the 2 copies of the repeat. A 21-bp FLT3-ITD is shown (17-bp c.1764_1780 duplication, with 4 additional N-nucleotides). These models are applicable to either simple replication slippage or break-induced models of MMRDR. The latter can occur following replication across a single-stranded nick and resulting replication fork collapse; fork collapse creates a single-ended double-stranded break, and the 3′ end must invade the template strand to reinitiate replication.
We asked whether identical ITDs existed, driven by longer microhomology blocks. The majority (52 of 72; 72%) of type A-D ITDs identified more than once in the cohort showed visible microhomology, with 17 different recurrent ITDs with visible microhomology observed (supplemental Table 4A). The 2 most highly recurrent were a 21-bp c.1780_1800dup driven by 4-bp TGAT microhomology, and a 21-bp c.1784-1804dup with 3-bp TCA microhomology. These 2 duplications account for 6% of all ITDs and create a spike at 21 bp in the FLT3-ITD length distribution (supplemental Figure 2). There is a positive correlation between recurrence and microhomology length (ρ = 0.991; P = <.0001; Spearman rank correlation) (supplemental Figure 3). Start position c.1770 was also unexpectedly associated with recurrence in the absence of microhomology (supplemental Table 4B) for reasons explored further under "Triggering the replication error." Few other microhomology block pairings >3 bp exist that could cause in-frame FLT3-ITDs between the type A/B start and end point ranges (supplemental Table 5). Visible germline microhomology is therefore an important, but not universal, determinant of FLT3-ITD genesis, with many of the potential longer blocks of microhomology used.
Filler junctions: identification of fillers as TdT-synthesized N-nucleotides
Insertion of filler nucleotides at the junction between the 2 repeats has been noted in approximately one-third of FLT3-ITDs.2,5,6,18,21 We reasoned that understanding the origin of these nucleotides might help decipher FLT3-ITD genesis.
Determining the origin of short insertions is not necessarily straightforward as such fragments may provide chance matches, and even a short insertion can originate from 2 or more separate tracts through sequential replication errors.22 We considered whether fillers had originated from exons 14 to 15 of FLT3, ±1-kb flanking DNA, aligning only fillers of ≥7 bp to reduce chance matches. Matches between 3 of 27 fillers were identified, each 7 to 8 bp, and attributed to chance. These results argue against a serial replication slippage origin within a single replication fork for the majority of filler sequences. Alternatively, the fillers could have originated elsewhere in the genome, through replication switching outside of the immediate replication fork or oligonucleotide capture.23 BLAST searches were performed using filler sequences >20 bp. Only 2 such fillers were available (28 bp and 36 bp), and neither was identified elsewhere in the genome as a single tract.
We alternatively considered whether the fillers might represent template-independent syntheses by a member of the polymerase X family, including TdT. TdT might seem an unlikely candidate as a myeloid mutagen, whereas both Pol μ and Pol λ are widely expressed; TdT is regarded as a lymphoid-specific enzyme. However, up to 55% of AML patients are known to be TdT+,24 and other patients may have downregulated TdT by diagnosis. Moreover, only TdT invariably polymerizes in a template-independent fashion. We present below 5 lines of evidence suggesting that TdT is responsible for filler synthesis, although a minor contribution from other enzymes cannot be excluded.
First, we considered the G/C content of the filler. TdT is biased toward addition of G and C nucleotides.25-28 The G/C percentage of N-nucleotides synthesized by TdT is typically 57% to 70%,25,29,30 whereas there is no increase in the G/C content of fillers originating though other mechanisms.25 For comparison, the G/C content of the human genome and FLT3 exon 14 are 41% and 38%, respectively. The G/C content of a total of 492 bp of FLT3-ITD filler nucleotides (from 95 ITDs) was 66.9% (supplemental Table 6), consistent with synthesis by TdT. This result also argues against involvement of Pol μ, which displays a preference for deoxythymidine triphosphate and deoxycytidine triphosphate.27
Second, we considered the length and size range of FLT3-ITD fillers. The mean length of N-regions from recombination activating gene (RAG)-mediated events is 3 to 6 nt,25,29,30 with a range of 1 to 13 nt at antigen receptor loci25,29 (supplemental Table 6). Longer N-nucleotide tracts (up to 21 nt) are occasionally reported at RAG-mediated events at loci other than IGH and TCR, including deletions of BTG1 in B-ALL.30 As TdT-mediated N-regions increase in length, they decrease in frequency.25 In contrast, filler fragments from other origins routinely exceed 13 nt in length, and show a distinct peak of 1-nt additions with a broadly flat distribution from 2 to 40 nt.25 Examination of 114 FLT3-ITD filler lengths showed a mean length of 5.6 nt with a range of 1 to 36 nt. Only 7 exceeded 13 nt. The distribution of filler lengths followed that expected for TdT-mediated N-nucleotides at antigen receptor loci, with a minimal number of longer tracts as observed at illegitimate targets (supplemental Figure 4).
Third, we examined dinucleotide content. Lacking a template, TdT stacks the incoming dNTP onto the existing base at the 3′OH, disposing to runs of homopurine or homopyrimidine.31 Purine-purine (RR) and pyrimidine-pyrimidine (YY) dinucleotides are therefore overrepresented in TdT syntheses at both legitimate31 and illegitimate32 RAG-mediated events. Among the 8 RR and YY dinucleotides, the 4 homopolymers (GG, AA, TT, and CC) are the most highly overrepresented.31 Conversely, RY and YR dinucleotides are underrepresented. We tested whether a total of 404 dinucleotides from FLT3-ITD fillers showed such biases. For example, G accounts for 33.7% of all FLT3-ITD filler nucleotides, and hence the dinucleotide GG would be expected at a frequency of 0.337 × 0.337 = 0.114. Approximately 46 occurrences would therefore be predicted within the 404 dinucleotides, but we observed a significantly higher figure of 80 (P < .001). Table 1 shows that 5 of 8 of the RR and YY dinucleotides were observed at a higher level than anticipated, 3 of them significantly (AA, CC, and GG; all homopolymers). Conversely, 7 of 8 of the RY and YR dinucleotides were present at a lower level than anticipated, 3 of them significantly (CA, CG, and GC). These results strongly implicate TdT.
As a control, we analyzed 327 genomic dinucleotides spanning FLT3 exons 14 to 15 (supplemental Table 7). Global genomic dinucleotide analysis in humans has previously shown a threefold to approximately fivefold depletion of CG dinucleotides due to deamination of 5-methylcytosine to thymine, and consequently smaller increases in both TG and CA dinucleotides.33 TA is also globally depleted for reasons that are unclear.33 In the control FLT3 dinucleotides, both CG and TA were significantly depleted as expected (supplemental Table 7). The 2 most highly overrepresented dinucleotides were TG and CA as predicted, but neither P value reached significance (supplemental Table 7). The significant depletion of CA from the FLT3-ITD filler dinucleotide data set contrasts to its overrepresentation in germline DNA, suggesting that the former has not been exposed to evolutionary time scales, consistent with neosynthesis by TdT.
Fourth, we compared the incidence of filler in FLT3-ITDs with the level of TdT positivity across the AML FAB types. Levels of TdT are highest in immature leukemias, especially FAB M0, and low overall in M3 (acute promyelocytic leukemia [APL]).24,34-36 FAB type was available for 211 FLT3-ITD+ AMLs with type A-D duplications (supplemental Table 8). The incidence of filler in all FAB types was 35% (74 of 211), but was significantly higher in M0 (10 of 12; 83%) (P = .0017; Fisher exact test; M0 against all other FAB types bar M3), and significantly lower in M3 (1 of 24; 4%) (P = .0009; Fisher exact test; M3 against all other FAB types bar M0), consistent with synthesis by TdT. In APL, the low level of filler in FLT3-ITDs is reflected by significantly higher use of visible microhomology (17 of 24 types A-D), particularly ≥2 bp (50% [12 of 24], cf 16% [29 of 187] in all other duplications; P = .0003, Fisher exact test). This results in a reduced palette of FLT3-ITDs in APL patients, slanted toward use of recurrent microhomology-driven ITDs. In the 24 APL duplications, the level of the recurrent 21-bp c.1780-1800dup reached 25% (6 of 24), and there were 3 examples of an 18-bp c.1790-1807dup driven by AAT microhomology.
Finally, we determined whether there was a higher incidence of filler in FLT3-ITDs in ALL compared with AML because ALL shows a higher level of TdT positivity.36,37 Filler incidence was 37% (104 of 284) and 63% (10 of 16) in AML and ALL, respectively (P = .061; Fisher exact test) (supplemental Table 2). This result did not reach significance, possibly due to the small number of lymphoid FLT3-ITDs. The sequences of a further 5 FLT3-ITDs from patients with ALL were obtained from a reference not initially identified.38 The revised data showed that 15 of 21 (71%) ALL ITDs had filler, significantly higher than myeloid FLT3-ITDs (P = .002).
Occult microhomology junctions
The data presented in the previous 2 sections suggest that TdT synthesizes the short junctional fillers, hereafter referred to as N-regions. We reasoned that TdT could also add 1 or more additional N-nucleotides, thereby creating the microhomology required for priming MMRDR. As these bases would by definition match the existing FLT3 sequence, their addition would not be apparent by inspection of the final sequence (occult microhomology).
Occult microhomology could also extend to those FLT3-ITDs lacking either visible germline microhomology or N-regions. Here, we envisage that TdT adds 1 or more nucleotides, but the bases fully match the target sequence and hence leave no apparent evidence of TdT involvement. This would allow all FLT3-ITDs to be attributed to MMRDR, satisfying the requirement for microhomology with either preexisting germline or polymerase-generated microhomology (Figure 2B-C).
The FLT3-ITD data set provides an opportunity to test for occult microhomology. Any bases synthesized by TdT and used as microhomology for priming should still show a G/C bias. This should manifest as a peak in the G/C content of the FLT3 sequence (when measured across multiple ITDs) at a limited number of bases immediately flanking the repeat junction (minimally positions +1 or −1) (supplemental Figures 5 and 6). Such a peak should be visible in ITDs showing N-nucleotide addition or lacking visible microhomology, and in an unknown proportion of those patients showing 1 bp of microhomology (as this homology may be present by chance, with a further occult base added by TdT). In contrast, ITDs showing 2 or more bases of visible germline microhomology might not be expected to show such a peak.
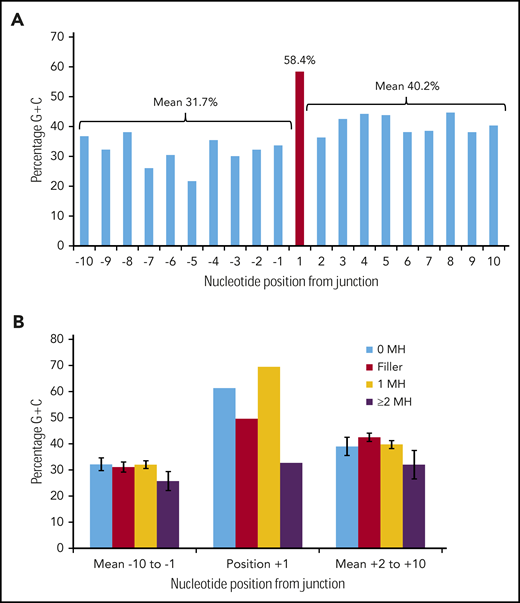
Figure 3A shows the G/C percentage across the 20 junction positions for 226 duplication FLT3-ITDs with either N-regions, no germline microhomology, or 1 bp of germline microhomology. There is a spike of 58.4% G/C at position +1, in contrast to means of 31.7% G/C for positions −10 to −1, and 40.2% for positions +2 to +10, consistent with the concept of occult microhomology (P = .003, Fisher exact test; position 1 vs 108-bp start region). In contrast, there was no evidence of a G/C spike at position +1 in 49 ITDs showing ≥2 bp of microhomology (Figure 3B). Neither set of results was affected by removal of repeat ITDs (data not shown). We do not attribute these results to a general requirement for G/C-rich microhomology priming, as the overall G/C content of the 1- to 6-nt germline microhomology was only 25.1% (supplemental Table 3). These data suggest that occult microhomology generated by TdT may prime at least 80% of all FLT3-ITDs, excluding only those primed by ≥2 bases of germline microhomology, and that a single base of occult microhomology will suffice.
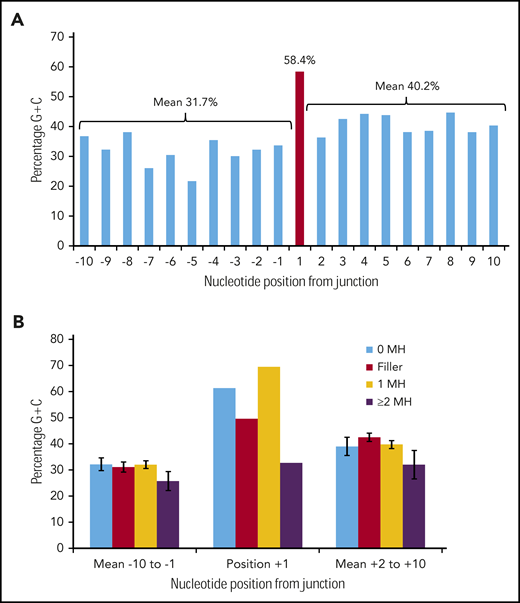
Occult microhomology G/C content fingerprint in FLT3-ITD junctions. (A) Percentage G/C content of the 20-nt positions flanking the FLT3-ITD repeat junction derived from 226 duplication junctions (N-nucleotide filler, n = 97; no visible microhomology, n = 70; 1 bp of microhomology, n = 59). Junctions with ≥2-bp microhomology were excluded, as their visible germline homology may be considered sufficient to prime MMRDR, and only a limited number of reintegration sites are available. A significant G/C spike is visible at position +1 (red). (B) Junction G/C content for individual junction categories. Error bars show standard error of the mean. There is no increase in G/C content for junctions with ≥2-bp microhomology (n = 49). MH, microhomology.
Occult microhomology G/C content fingerprint in FLT3-ITD junctions. (A) Percentage G/C content of the 20-nt positions flanking the FLT3-ITD repeat junction derived from 226 duplication junctions (N-nucleotide filler, n = 97; no visible microhomology, n = 70; 1 bp of microhomology, n = 59). Junctions with ≥2-bp microhomology were excluded, as their visible germline homology may be considered sufficient to prime MMRDR, and only a limited number of reintegration sites are available. A significant G/C spike is visible at position +1 (red). (B) Junction G/C content for individual junction categories. Error bars show standard error of the mean. There is no increase in G/C content for junctions with ≥2-bp microhomology (n = 49). MH, microhomology.
Triggering the replication error
A 30-bp imperfect palindrome (c.1778-1807, centered 1792/3) was previously suggested to promote FLT3 replication errors.16 Our data set showed no evidence of overall increased use of the palindrome start, hairpin tip, or end points (supplemental Table 9; supplemental Figure 7). Moreover, ITD start and end points were both found within or either side of the palindrome (supplemental Table 9).
We show herein that the majority (52 of 72) of recurrent ITDs are driven by germline microhomology. We further reasoned that some of the remaining recurrent ITDs lacking germline microhomology might instead relate to secondary structure responsible for triggering replication slippage. Notably, a group of 12 ITDs all shared a common start point of c.1770, coupled to 1 of 3 end points (c.1793, n = 7; c.1811, n = 3; and c.1830, n = 2) (supplemental Table 4B). Importantly, the c.1793 end point corresponds to the tip of the c.1778-1807 palindrome previously identified.16 However, positions 1770 and 1811 do not correspond to the palindrome start and end points. We therefore propose an extended c.1770_1812 palindrome (Figure 4). In this structure, the recurrent c.1770_1793dup and c.1770_1811dup ITDs correspond to start → hairpin tip and start → end duplications (Figure 4).

Original and revised palindromes proposed to destabilize FLT3 exon 14 and trigger MMRDR. (A) The 30-bp c.1778-1807 imperfect palindrome originally proposed. (B) The revised c.1770_1812 structure proposed here. The recurrent c.1770_1793dup ITD corresponding to start → hairpin tip is delineated in blue, and the recurrent c.1770_1811dup start → end duplication in red. FLT3-ITDs start before or within this revised structure, and end within or after, with only rare exceptions.
Original and revised palindromes proposed to destabilize FLT3 exon 14 and trigger MMRDR. (A) The 30-bp c.1778-1807 imperfect palindrome originally proposed. (B) The revised c.1770_1812 structure proposed here. The recurrent c.1770_1793dup ITD corresponding to start → hairpin tip is delineated in blue, and the recurrent c.1770_1811dup start → end duplication in red. FLT3-ITDs start before or within this revised structure, and end within or after, with only rare exceptions.
The significance of this structure was further assessed through comparison of the 278 FLT3-ITD type A-D start and end points to positions c.1770 and c.1812 (supplemental Table 9). One hundred forty-four of 278 start points (51.8%) occurred prior to the extended palindrome and the remainder within the palindrome. The last start position corresponded exactly to the last base of the palindrome, and no start points were observed after this position. A comparable converse pattern was observed for the end points; 2 of 278 (0.7%), 158 of 278 (56.8%), and 118 of 278 (42.4%) were observed before, within, and after the extended palindrome, respectively. The 2 end points found before the palindrome were close to the start point. Fifty of 278 ITDs (18.0%) started before and ended after the palindrome, showing that many ITDs span the palindrome, and that a breakpoint within the palindrome is not required. FLT3-ITDs, therefore, almost invariably start before or within the revised palindrome and end within or after the revised palindrome, confirming the significance of this structure. We suggest that this palindrome triggers MMRDR, with misalignment promoted by TdT.
Discussion
The genetic landscape of AML includes many cytogenetic and molecular lesions now exploited for diagnostic, prognostic, monitoring, and therapeutic purposes.39 Understanding how these mutations occur is also critical. For example, the recurrent translocations and inversions in AML arise following chromosome breakage and NHEJ-mediated repair. However, the breakage sites are not necessarily random, and in therapy-related AML often occur at topoisomerase II–binding sites.40 Analysis of base substitutions in AML has identified just 2 causative mutational signatures, deamination of 5-methyl-cytosine, reflecting age,41 and the ubiquitous mutational signature 5.42 Such studies have left the genesis of FLT3-ITDs unresolved.
Here, we explore the origin of FLT3-ITDs in AML. We identify an unexpected role for TdT, proposing that the majority of FLT3-ITDs occur following addition of N-nucleotide(s) by TdT during MMRDR. The physiological function of TdT is to add N-nucleotides to single-stranded DNA during V(D)J recombination to enhance antigen receptor diversity.28 This ability to synthesize DNA in the absence of a template strand is unusual among polymerases,28 with a clear potential for off-target activity to cause neoplasia. However, TdT mutagenesis has not previously been identified as carcinogenic. Expression of TdT is essentially restricted to lymphoid cells to help limit illegitimate activity, but its frequent expression in myeloid stem cells may risk the development of AML. DNA processes requiring extension from a 3′ end, such as MMRDR, may be exquisitely sensitive to TdT activity as the 3′ tip is critical for alignment.43 TdT is believed to have ready access to free DNA ends in the nucleus.44
We did not set out to implicate TdT in leukemogenesis, but instead explore how FLT3-ITDs were primed. We initially identified 1 to 6 bp of germline microhomology suitable for priming a minority of ITDs. To explain how the remaining ITDs are primed, we propose that TdT adds short runs of N-nucleotides to the 3′ end of the misaligning strand. The last nucleotide added is used for priming (occult microhomology), and any previous nucleotides appear as N-regions at the duplication junction. We suggest that this activity by TdT both permits priming at an incorrect site and inhibits alignment at the correct site. We support this model by identifying the unique footprint of TdT neosynthesis at FLT3-ITD repeat junctions. This footprint, conferred by the unusual properties of this polymerase, is most easily visualized within the N-regions. TdT’s bias toward addition of G and C nucleotides results in an elevated G/C content, whereas its lack of a template creates a predisposition toward nucleotide stacking, resulting in a uniquely skewed dinucleotide composition. We are unaware of any other polymerases capable of similar nontemplated syntheses. Moreover, analysis of junction sequences from BCOR-ITDs (duplications of comparable size found in specific solid tumors that lack expression of TdT45 ) failed to reveal equivalent G/C-rich insertions (J.B., unpublished data, 10 August 2019). Additional to the N-region analysis, we identify a G/C-rich spike representing occult microhomology at a position immediately adjacent to the duplication junction. Furthermore, our results do not exclude the possibility that some ITDs apparently primed by germline microhomology still occur following N-nucleotide addition by TdT, as such addition would still inhibit correct realignment. The failure to detect a G/C-rich spike at position +1 in ITDs with germline microhomology of ≥2 bases may reflect the limited availability of matching sites.
The occurrence of multiple FLT3-ITDs in a single patient suggests that FLT3 exon 14 is prone to rearrangement. This may represent both the destabilizing effect of the extended palindrome and the action of TdT. Unselected out-of-frame FLT3-ITDs are also expected to occur. Although the effect of the palindrome may be uniform, progenitor cells transforming with high TdT might be predicted to have a high FLT3-ITD incidence, with an increased proportion showing N-regions. We support our model by showing an association across FAB types between TdT levels and the incidence of FLT3-ITD N-regions. This increase is also seen in FLT3-ITDs from ALL, where TdT levels are markedly high.
The varying incidence of FLT3-ITDs across AML cytogenetic types allows further examination of this hypothesis. The overall incidence of FLT3-ITDs in AML is 25%, but lower (7% to 9%) in patients with t(8;21) or inv(16), and lower again (2% to 4%) in patients with a complex karyotype.10,11 In contrast, 90% of patients with t(6;9) DEK-NUP2143,42 or t(5;11) NUP98-NSD146,47 are FLT3-ITD+. These differences could reflect preferential cooperation between mutations and/or differential activity of a mutator. Our model suggests that t(6;9) and t(5;11) stem cells might express significant levels of TdT. The t(6;9) is indeed recognized to arise from an early hematopoietic precursor associated with high levels of TdT,48 although the TdT status of t(5;11) leukemias is unknown. Furthermore, in t(15;17) PML-RARA (APL), the incidence of FLT3-ITDs is 35% overall,9,10 but ranges from 23% in hypergranular M3 to 65% in the rarer hypogranular M3v.10 Hypergranular APL arises from a myeloid committed progenitor and typically lacks lymphoid antigens,49 whereas M3v arises from an earlier CD34+ progenitor and often coexpresses lymphoid antigens.49-52 TdT is only rarely detected in hypergranular M3, but more commonly in M3v.51-53 These data are consistent with the concept that M3v cases occur in a progenitor with high TdT and are more likely to acquire a FLT3-ITD. As the distinction between M3 and M3v was not clear throughout our cohort, this idea requires confirmation. Overall, APL patients may show a higher reliance on germline microhomology.
In our accompanying manuscript, we confirm and extend this AML TdT-mutator model to the genesis of NPM1 mutations, which we also propose require priming by TdT.54 We suggest that TdT may be a significant cause of AML, and that additional examples of TdT mutagenesis in select neoplasms will emerge.
Data may be found in supplemental Figures 1-7 and supplemental Tables 1-9.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank David Schatz and Joydeep Banerjee (School of Medicine, Yale University) for helpful discussions, and Joanne Mason (West Midlands Regional Genetics Laboratory) for critical reading of the manuscript.
Authorship
Contribution: J.B. conceived the study, assembled the FLT3-ITD cohort, analyzed data, performed statistical analyses, and wrote the draft manuscript; S.A.D., S.A., and M.J.G. supervised the project; and all authors provided intellectual input and revised and gave final approval to the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Julian Borrow, West Midlands Regional Genetics Laboratory, Birmingham Women’s and Children’s NHS Foundation Trust, Mindelsohn Way, Edgbaston, Birmingham, B15 2TG, United Kingdom; e-mail: j.borrow@nhs.net.