

In this issue of Blood, have uncovered a surprising role of neutrophil extracellular traps (NETs) as a potent driver of inflammatory cytokine production in an autoinflammatory disease called deficiency of adenosine deaminase 2 (DADA2).1
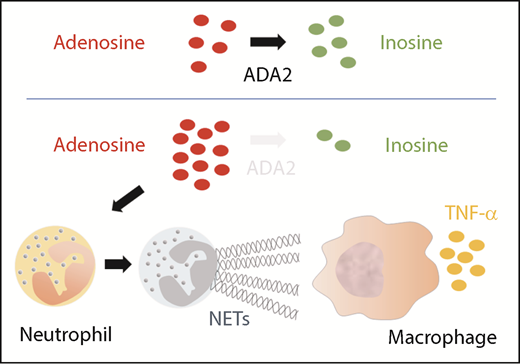
Deficiency of ADA2 triggers NET formation, which activates macrophages to produced TNF-α.
Deficiency of ADA2 triggers NET formation, which activates macrophages to produced TNF-α.
DADA2 is a fascinating monogenic disorder caused by biallelic mutations in adenosine deaminase 2 (ADA2; formerly known as CECR1) on chromosome 22q11. Initially described in 2014,2,3 about 200 cases have now been published in the literature to date. DADA2 is primarily characterized by childhood-onset inflammatory vasculopathy, early-onset strokes, bone marrow failure, and immunodeficiency. However, disease manifestations and severity vary considerably among patients, even in those with identical genotype. Accumulating experience in the field suggests a crucial role of the proinflammatory cytokine tumor necrosis factor α (TNF-α) in this disease because TNF inhibitors can effectively control the inflammatory vasculopathy and prevent further strokes in DADA2 patients.4 Proinflammatory M1 monocytes and macrophages are thought to be the source of TNF-α, but how TNF-α production is triggered in the absence of ADA2 is an important question that remains unanswered. Carmona-Rivera and colleagues found that NET formation by neutrophils (also called NETosis) may be the missing link that translates ADA2 deficiency into macrophage activation and subsequent TNF-α production.
To understand the proposed mechanism, let us quickly revisit the biology of the human ADA enzyme family. Adenosine is a purine nucleoside that can signal multiple biological pathways by differential activation of adenosine receptors.5 Whereas both ADA1 and ADA2 catalyze the deamination reaction that converts adenosine to inosine (see figure), the 2 enzymes are not functionally redundant. ADA1 is an intracellular monomeric protein produced ubiquitously, and mutations in ADA1 result in severe combined immunodeficiency resulting from an accumulation of toxic metabolites in developing lymphocytes. In contrast, ADA2 is an extracellular homodimer secreted by monocytes and macrophages, but whether it regulates extracellular adenosine homeostasis at physiologic levels of adenosine is a subject of debate.6 ADA2 may possess additional functions, given its wide spectrum of clinical manifestations in DADA2.
The formation of NETs by neutrophils is vital to innate immunity against microbes, but it also promotes inflammation in autoimmune diseases such as vasculitis.7 The work by Carmona-Rivera et al provides intriguing in vitro evidence that an accumulation of extracellular adenosine in the absence of ADA2 can promote NET formation. DADA2 patients not only have higher levels of plasma adenosine but also have increased numbers of circulating low-density granulocytes (LDGs) that are primed to undergo NETosis. Supporting these findings, NETs were abundant in the peripheral blood and affected tissue from DADA2 patients with active disease. Importantly, incubation with NETs from DADA2 patients induced M1 macrophages to produce large amounts of TNF-α, thereby providing a novel mechanistic link that couples defective ADA2 function with inflammatory cytokine production. Circulating LDGs in DADA2 patients were significantly reduced during disease remission, suggesting a possible feedback loop between inflammation and NETosis in this disease.
Currently, most DADA2 patients with the vasculopathy phenotype are treated with biologics that target TNF-α, whereas bone marrow transplantation provides a curative option for patients with refractory disease or severe bone marrow failure.4,8 With a better understanding of the molecular events upstream of TNF-α production, we may uncover additional therapeutic options for DADA2. To further pursue the mechanism of NETosis in DADA2, Carmona-Rivera and colleagues determined the requirement of adenosine receptors A1 and A3, reactive oxygen species, and peptidylarginine deiminase for adenosine-induced NETosis. Although it is not clear how NETs activate macrophages, agonistic activation of adenosine receptor A2A downregulated NET-induced secretion of TNF-α by macrophages. As we continue to unravel the physiologic function of ADA2 and the pathophysiology of DADA2, we will need studies that evaluate the therapeutic potentials of targeting adenosine receptors and downstream signal pathways.
The heterogeneity of DADA2-associated disease manifestations should be kept in mind because the Carmona-Rivera study focuses primarily on patients with inflammatory vasculopathy.9,10 Cytopenias with a full spectrum of severity, including neutropenia that can be chronic and refractory to immunosuppression, have been described in DADA2 patients with or without inflammatory vasculopathy. This suggests that neutrophil-independent pathways may also contribute to the inflammatory response, although neutrophil abundance and NETosis at the tissue level are not assessed by routine laboratory studies. Whether NETosis and NET-induced TNF-α also occur in DADA2 patients with bone marrow failure or immunodeficiency as primary disease manifestation will require further evaluation.
Conflict-of-interest disclosure: The author declares no competing financial interests.

