

Abstract
Since its discovery, polycythemia vera (PV) has challenged clinicians responsible for its diagnosis and management and scientists investigating its pathogenesis. As a clonal hematopoietic stem cell (HSC) disorder, PV is a neoplasm but its driver mutations result in overproduction of morphologically and functionally normal blood cells. PV arises in an HSC but it can present initially as isolated erythrocytosis, leukocytosis, thrombocytosis, or any combination of these together with splenomegaly or myelofibrosis, and it can take years for a true panmyelopathy to appear. PV shares the same JAK2 mutation as essential thrombocytosis and primary myelofibrosis, but erythrocytosis only occurs in PV. However, unlike secondary causes of erythrocytosis, in PV, the plasma volume is frequently expanded, masking the erythrocytosis and making diagnosis difficult if this essential fact is ignored. PV is not a monolithic disorder: female patients deregulate fewer genes and clinically behave differently than their male counterparts, while some PV patients are genetically predisposed to an aggressive clinical course. Nevertheless, based on what we have learned over the past century, most PV patients can lead long and productive lives. In this review, using clinical examples, I describe how I diagnose and manage PV in an evidence-based manner without relying on chemotherapy.
Introduction
Polycythemia vera (PV) is the commonest myeloproliferative neoplasm (MPN), the ultimate phenotypic consequence of JAK2 somatic driver mutations, and the MPN most often complicated by arterial and venous thrombosis because it is the only one in which erythrocytosis occurs. First recognized in 1892, PV has been studied for 125 years and, despite its infrequency, it has captured the imagination of physicians in every generation. Osler explained the reason eloquently:
Nothing is more certain—in the microcosm as in the macrocosm, given a demand and there is soon a supply. But here is a condition in which, so far as we know, there is an over-supply without any corresponding demand and the same riddle confronts us as in leukemia and several other diseases of which over-production of a normal tissue or element is of the essence.1 (p145)
Such fascination has consequences. Since the 2014 Blood article “How I treat polycythemia vera,”2 there have been 589 publications about PV diagnosis and 655 about its management; these numbers exceed the ability of the most efficient practitioners to critically digest. Yet, despite such scholarly activity, there is no consensus on how to diagnose PV,3,4 how to manage it proactively as opposed to supportively, or whether PV is a separate entity from essential thrombocytosis (ET),5,6 even though they are genetically distinct diseases.7,8 Although the molecular basis of PV remains elusive, there is no reason for this situation to persist. There are sufficient clinical and scientific data to diagnose PV with almost complete accuracy,9 and to develop evidence-based treatment guidelines that embrace the dictates of precision medicine.10 In this review, I describe an approach to the diagnosis and management of PV using informative patients. Previous articles have comprehensively reviewed the relevant literature before the discovery of JAK2V617F 11 , and since its discovery12 (the latter both through its references and those in its supplemental data).
Hematopoietic stem cell biology
The 3 MPNs (PV, ET, and primary myelofibrosis [PMF]) are clonal hematopoietic stem cell (HSC) disorders; understanding HSC biology is critical to MPN diagnosis and management. Figure 1A illustrates the current concept of the HSC hierarchy.13 Importantly, in addition to the long-term HSC (LT-HSC)–short-term HSC (ST-HSC) pathway, LT-HSCs give rise directly to megakaryocytic-repopulating (MkRP), megakaryocytic and erythroid–repopulating (MERP), and common myeloid–repopulating (CMRP) HSCs. From this perspective, there are actually 3 HSC hierarchies: an LT-HSC–repopulating cell hierarchy, a hematopoietic growth factor receptor hierarchy with MPL at its apex, and a proliferative hierarchy with a myeloaccumulative apex and a myeloproliferative base. These 3 hierarchies define the origin of the MPN, a basis for the transitions between them (Figure 1B-C), and the differential sensitivity of HSCs and their committed progenitor cell progeny to targeted therapy.
![Figure 1. HSC behavior in PV. (A) HSC physiology. Current concepts of the HSC hierarchy.13 At the apex of the HSC hierarchy is the long-term HSC (LT-HSC; CD34+CD38− cell), which is responsible for lifetime maintenance of bone marrow cellular integrity and in which all MPN driver mutations are expressed. In addition to the classical commitment pathway through the short-term HSC (ST-HSC), LT-HSCs can give rise directly to committed, self-repopulating HSCs restricted to megakaryocytopoiesis (MkRP), megakaryocytopoiesis and erythropoiesis (MERP), or all myeloid cells (common myeloid–repopulating [CMRP]). Importantly, the thrombopoietin receptor, MPL, is the only hematopoietic growth factor receptor expressed in LT-HSCs because, in addition to its effects as an HSC growth factor, it is responsible for tethering LT-HSCs in their marrow niches to osteoblast-expressed thrombopoietin (THPO). LT-HSCs are largely dormant, insensitive to MPN driver mutations; expansion of the MPN LT-HSC population at the expense of normal LT-HSCs can take years to occur. At the base of the hierarchy are the committed hematopoietic progenitor cells, which are hyperproliferative, addicted to MPN driver mutations, and sensitive to JAK1/2 inhibitors. (B) Conversion to PV in an ET patient 6 years after diagnosis associated with the development of JAK2V617F homozygosity, a PV hallmark. (C) Conversion to PV in a PMF patient after 17 years. The bar indicates hydroxyurea therapy. EPO-R, erythropoietin receptor; ery, erythroid progenitor; G-CSFR, granulocyte colony-stimulating factor receptor; LMPP, lymphoid-primed multipotent progenitor; meg, megakaryocyte; MP, myeloid progenitor; MyRP, myeloid-restricted repopulating progenitor; n/m, neutrophil/monocyte progenitor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/4/10.1182_blood.2018834044/3/m_bloodbld2018834044cf1.png?Expires=1768490811&Signature=d6kwuQPAYYhwQanhd~-SK3hb0kt65jyjDBd8e46bBd7d-JFhsO4Cx1vsDZkhQWYSDMkfxjYW2ewqnhDz2qTRIUjWM8pjhMc-yAMqgKQbcMWlcG65JfaKtJv2PSukd82t~jj9ZxGW65-zUfH2saCnTIE7rRNRNM-MvsRGsUFP5LrNljUmoT8jcjQf8mD4nyIIBgLT~GG9AmUuJ3RZAcizYX8MzyRl75GWuNHBrj7fDfUzyYLveF27m-EQBTK1p7v2kHEoxmkiCS-4Coqfx~cEsajuYVYL-LK~XTfyHVpSX~0nXlqIXrzETIyINHWxNZeY9Y1QBH3XsuOi9ejBr6Q-LA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HSC behavior in PV. (A) HSC physiology. Current concepts of the HSC hierarchy.13 At the apex of the HSC hierarchy is the long-term HSC (LT-HSC; CD34+CD38− cell), which is responsible for lifetime maintenance of bone marrow cellular integrity and in which all MPN driver mutations are expressed. In addition to the classical commitment pathway through the short-term HSC (ST-HSC), LT-HSCs can give rise directly to committed, self-repopulating HSCs restricted to megakaryocytopoiesis (MkRP), megakaryocytopoiesis and erythropoiesis (MERP), or all myeloid cells (common myeloid–repopulating [CMRP]). Importantly, the thrombopoietin receptor, MPL, is the only hematopoietic growth factor receptor expressed in LT-HSCs because, in addition to its effects as an HSC growth factor, it is responsible for tethering LT-HSCs in their marrow niches to osteoblast-expressed thrombopoietin (THPO). LT-HSCs are largely dormant, insensitive to MPN driver mutations; expansion of the MPN LT-HSC population at the expense of normal LT-HSCs can take years to occur. At the base of the hierarchy are the committed hematopoietic progenitor cells, which are hyperproliferative, addicted to MPN driver mutations, and sensitive to JAK1/2 inhibitors. (B) Conversion to PV in an ET patient 6 years after diagnosis associated with the development of JAK2V617F homozygosity, a PV hallmark. (C) Conversion to PV in a PMF patient after 17 years. The bar indicates hydroxyurea therapy. EPO-R, erythropoietin receptor; ery, erythroid progenitor; G-CSFR, granulocyte colony-stimulating factor receptor; LMPP, lymphoid-primed multipotent progenitor; meg, megakaryocyte; MP, myeloid progenitor; MyRP, myeloid-restricted repopulating progenitor; n/m, neutrophil/monocyte progenitor.
HSC behavior in PV. (A) HSC physiology. Current concepts of the HSC hierarchy.13 At the apex of the HSC hierarchy is the long-term HSC (LT-HSC; CD34+CD38− cell), which is responsible for lifetime maintenance of bone marrow cellular integrity and in which all MPN driver mutations are expressed. In addition to the classical commitment pathway through the short-term HSC (ST-HSC), LT-HSCs can give rise directly to committed, self-repopulating HSCs restricted to megakaryocytopoiesis (MkRP), megakaryocytopoiesis and erythropoiesis (MERP), or all myeloid cells (common myeloid–repopulating [CMRP]). Importantly, the thrombopoietin receptor, MPL, is the only hematopoietic growth factor receptor expressed in LT-HSCs because, in addition to its effects as an HSC growth factor, it is responsible for tethering LT-HSCs in their marrow niches to osteoblast-expressed thrombopoietin (THPO). LT-HSCs are largely dormant, insensitive to MPN driver mutations; expansion of the MPN LT-HSC population at the expense of normal LT-HSCs can take years to occur. At the base of the hierarchy are the committed hematopoietic progenitor cells, which are hyperproliferative, addicted to MPN driver mutations, and sensitive to JAK1/2 inhibitors. (B) Conversion to PV in an ET patient 6 years after diagnosis associated with the development of JAK2V617F homozygosity, a PV hallmark. (C) Conversion to PV in a PMF patient after 17 years. The bar indicates hydroxyurea therapy. EPO-R, erythropoietin receptor; ery, erythroid progenitor; G-CSFR, granulocyte colony-stimulating factor receptor; LMPP, lymphoid-primed multipotent progenitor; meg, megakaryocyte; MP, myeloid progenitor; MyRP, myeloid-restricted repopulating progenitor; n/m, neutrophil/monocyte progenitor.
Natural history
The natural history of PV is ill-defined because of phenotypic mimicry with ET and PMF,2 and, as Dameshek noted:
It is difficult to state what the normal course of the disease would be without the various therapeutic methods which undoubtedly influence it.14(p793)
Traditionally, PV was thought to progress serially from erythrocytosis to myelofibrosis and leukemic transformation,15 but its presentation and progression are really functions of host genetic variation12 including sex and age16 ; an alternate disease model is illustrated in Figure 2. Gene-expression profiling established that PV is not a monolithic disorder but occurs in indolent and aggressive forms, and the latter has a unique gene signature8 ; recent studies also suggest that deleterious mutation accumulation might identify patients at risk of disease transformation.17-19
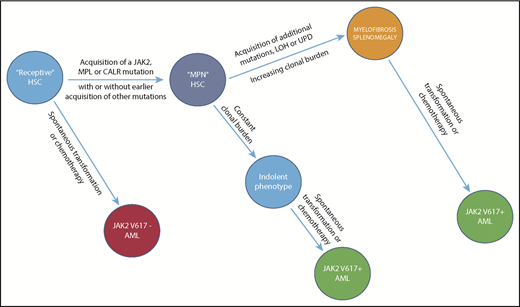
Evolution of a myeloid neoplasm. The natural history of JAK2V617F-positive PV, illustrating the evolution of subclones (only 1 is shown) from the founding LT-HSC clone, which first acquires JAK2V617F and, with time, additional fitness mutations, though the latter are not mandatory for clonal expansion. Importantly, leukemic transformation can occur at several levels. It is JAK2V617F-negative when arising in the founding LT-HSCs, and JAK2V617F-positive when arising from an involved LT-HSC daughter clone. The acquisition and phenotypic consequences of these mutations appear to be due to and modified not only by host genetic variation, in particular, age and sex, but also by chemotherapy exposure. Clonal dominance, in which there is suppression of normal LT-HSCs by the malignant clone, is a central feature of PMF and, to a lesser extent and later in its natural history, of PV, usually resulting in leukocytosis and extramedullary hematopoiesis. Clonal dominance is not a function of a particular MPN driver mutation but rather, a function of the MPN driver mutation VAF frequency at the level of the involved LT-HSC; it is usually present when the neutrophil allele burden is ≥70%.59 LOH, loss of heterozygosity; UPD, uniparental disomy.
Evolution of a myeloid neoplasm. The natural history of JAK2V617F-positive PV, illustrating the evolution of subclones (only 1 is shown) from the founding LT-HSC clone, which first acquires JAK2V617F and, with time, additional fitness mutations, though the latter are not mandatory for clonal expansion. Importantly, leukemic transformation can occur at several levels. It is JAK2V617F-negative when arising in the founding LT-HSCs, and JAK2V617F-positive when arising from an involved LT-HSC daughter clone. The acquisition and phenotypic consequences of these mutations appear to be due to and modified not only by host genetic variation, in particular, age and sex, but also by chemotherapy exposure. Clonal dominance, in which there is suppression of normal LT-HSCs by the malignant clone, is a central feature of PMF and, to a lesser extent and later in its natural history, of PV, usually resulting in leukocytosis and extramedullary hematopoiesis. Clonal dominance is not a function of a particular MPN driver mutation but rather, a function of the MPN driver mutation VAF frequency at the level of the involved LT-HSC; it is usually present when the neutrophil allele burden is ≥70%.59 LOH, loss of heterozygosity; UPD, uniparental disomy.
PV can occur at any age because JAK2V61F expression is age independent,20 but it occurs at a younger age in women and its frequency increases exponentially after the age of 60 years21 with male predominance, when the frequency of clonal hematopoiesis of indeterminate potential22 and acute myelogenous leukemia (AML) in the general population increase.21 PV disease complications differ with sex23 but not age24,25 though they are aggravated by age26 because aging, not disease duration, is associated with accumulation of potentially harmful mutations,22 which might in part be related to promotion of genetic instability by JAK2V617F.27 Disease duration correlates with myelofibrotic transformation,8,28 but not in all patients.29 What has been characterized as “spent-phase” PV30 is not bone marrow failure but rather a combination of iron deficiency, splenomegaly, plasma volume expansion, and disease duration.
The MPNs are dynamic, and subject to transformations among themselves due to shared driver mutations, making MPN diagnosis a moving target. For example, JAK2V617F-positive ET can transform to PV (Figure 1B), more often in women,16 as can JAK2V617F-positive PMF (Figure 1C). This behavior may reflect involvement of a different HSC clone (Figure 1A); clonal expansion due to JAK2V617F homozygosity, a feature of PV31 ; or clinical recognition of masked PV.32,33
Approximately 10% of patients develop “post-PV myelofibrosis” (PPMF),34 an unfortunate appellation conjuring comparison with PMF, however, PPMF behaves differently.35 Marrow reticulin fibrosis is a reactive, reversible histologic process,36 which does not affect bone marrow function34 and can be present in PV at diagnosis without impacting survival.37,38
Leukemic transformation, the most serious PV consequence, can develop in chronic phase, but more frequently during PPMF, spontaneously or associated with chemotherapy or irradiation,39,40 particularly in patients age ≥60 years. Neither JAK2V617F expression8 nor its variant allele frequency (VAF)18 correlate with leukemic transformation, genomic changes,41 or survival, unless there is mutation homozygosity.42 Approximately 30% of cases are JAK2V617F-negative, presumably originating in the ancestral LT-HSC clone43,44 (Figures 1A and 2); recent studies suggest genetic instability is less marked in PPMF than in PMF.45
Diagnosis
Recognizing PV from the panoply of diseases it mimics is not always easy. PV is a panmyelopathy: when it presents with erythrocytosis, leukocytosis, and thrombocytosis with or without splenomegaly, the diagnosis is confirmed, regardless of the clonal marker. PV, however, can present as isolated erythrocytosis, leukocytosis,46 thrombocytosis,33 or splenomegaly,31 with myelofibrosis, or any combination of these (Table 1).32,47,48 JAK2 driver mutation expression eliminates the possibility of secondary or spurious erythrocytosis but it does not distinguish PV from ET or PMF.
The variable presentation of PV
| PVSG, % | Sweden | |
|---|---|---|
| Erythrocytosis alone | 0 | 17 |
| Erythrocytosis and | ||
| Leukocytosis | 13 | 29 |
| Thrombocytosis | 30 | 16 |
| Leukocytosis and thrombocytosis | 57 | 38 |
| Splenomegaly (palpable) | 70 | 58 |
| Splenomegaly and | ||
| Leukocytosis | ND | 66 |
| Thrombocytosis | ND | 54 |
| PVSG, % | Sweden | |
|---|---|---|
| Erythrocytosis alone | 0 | 17 |
| Erythrocytosis and | ||
| Leukocytosis | 13 | 29 |
| Thrombocytosis | 30 | 16 |
| Leukocytosis and thrombocytosis | 57 | 38 |
| Splenomegaly (palpable) | 70 | 58 |
| Splenomegaly and | ||
| Leukocytosis | ND | 66 |
| Thrombocytosis | ND | 54 |
ND, not determined; PVSG, Polycythemia Vera Study Group.
The 2016 World Health Organization (WHO) PV diagnostic criteria addressed this problem, stipulating specific hemoglobin, hematocrit, and red cell mass (RCM) values, marrow histologic criteria, and a low serum erythropoietin.4 However, RCM assays are not widely available and the specificity of marrow histology as a PV diagnostic test has been challenged,3,49‐51 as has the value of the serum erythropoietin assay.3,50 Importantly, there can be discrepancies between hemoglobin and hematocrit values,50 and the stipulated thresholds do not address PV patients with masked erythrocytosis due to plasma volume expansion,12,32 an important feature in women, who are prone to hepatic vein thrombosis (HVT).23 The following 2 patients exemplify these diagnostic issues and suggest a solution.
Patient 1
An 86-year-old woman was referred for management of JAK2V617F-positive ET of 2 years’ duration. She was taking 81 mg of aspirin daily and was hydroxyurea-intolerant. She complained of anorexia, weight loss, fatigue, headaches, and erythromelalgia. Physical examination was normal; levels and counts were as follows: hematocrit, 0.45; hemoglobin gram percentage, 14.3 g/dL; red blood cells (RBCs), 5.6 × 1012/L (normal, 5.2 × 1012/L); mean corpuscular volume (MCV), 80 fL; absolute neutrophil count (ANC), 7.1 × 109 /L; and platelets, 138 × 109/L. The blood smear showed microcytes and elliptocytes. The JAK2V617F VAF was 55%. The elevated RBCs, borderline MCV, blood smear, and JAK2V617F VAF >50% suggested PV,16 which was confirmed by an RCM/plasma volume study showing an excess of 500 mL of erythrocytes, masked by 1 L of excess plasma.
Patient 2
A 60-year-old man had an ANC of 21.1 × 106/L; the other blood counts were normal. The marrow was hypercellular with myeloid predominance with full maturation and megakaryocyte hyperplasia but no increase in reticulin. Flow cytometry and cytogenetics were normal as was a BCR-ABL assay; a JAK2V617F assay was positive. Marrow examination at age 68 years revealed myeloid hyperplasia with full maturation, megakaryocytic hyperplasia, and grade 2 reticulin fibrosis. The JAK2V617F VAF was 86% and a serum erythropoietin level was <1 mU/mL. The spleen was 7 cm below the left costal margin. A diagnosis of PMF was made. When seen at Johns Hopkins, the patient’s hemoglobin gram percentage was 15.3 g/dL, hematocrit 0.52, RBCs were 6.8 × 1012/L, the MCV was 75.9 fL, the ANC was 43 × 109/L with a normal differential with nucleated red cells present, platelets were 152 × 109/L, and the reticulocyte count was 2.2%. The JAK2V617F VAF was 100%. On physical examination, there was plethora and massive splenomegaly. Microcytic erythrocytosis, particularly with splenomegaly, suggested PV rather than PMF. An RCM/plasma volume study was performed, documenting the presence of 1.7 L of excess erythrocytes and 498 mL of excess plasma, indicating masked PV.
In Osler’s era, the RBC count was recognized as a more accurate indicator of erythrocytosis than the hemoglobin level.52 These 2 patients illustrate this principle. When the supply of iron or globin is limited, the erythrocyte defends the mean corpuscular hemoglobin concentration at the expense of the MCV.53 Microcytic erythrocytosis is a hallmark of disorders causing erythrocytosis and thalassemia trait54 ; the hematocrit can also be affected50 as indicated in Figure 3, making the RBC the preferred marker of erythrocytosis in PV for diagnostic and therapeutic purposes if the hematocrit cannot be used due to the effects of iron deficiency or plasma volume expansion (Figure 4).

Comparison of the changes in the RBC, hemoglobin, and hematocrit over time in a PV patient treated only with phlebotomy, illustrating the variable quantitative impact of iron deficiency on these 3 measurements of erythropoiesis. The parallel bars indicate the upper (blue) and lower (green) limits of normal.
Comparison of the changes in the RBC, hemoglobin, and hematocrit over time in a PV patient treated only with phlebotomy, illustrating the variable quantitative impact of iron deficiency on these 3 measurements of erythropoiesis. The parallel bars indicate the upper (blue) and lower (green) limits of normal.
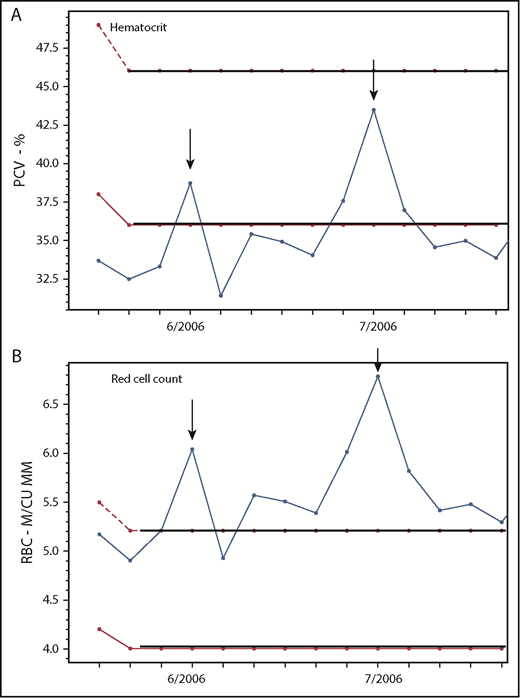
In PV, the red cell count is frequently a better indicator of erythrocytosis than the hematocrit. Changes in the (A) hematocrit and (B) RBC count in a 37-year-old woman with PV-associated HVT treated with phlebotomy, Coumadin, and a transjugular intrahepatic portosystemic shunt (TIPS), demonstrating the correlation of the RBC but not the hematocrit with TIPS thrombosis (arrows) on 2 occasions. M/CU MM, millions per cubic millimeter; PCV, packed cell volume.
In PV, the red cell count is frequently a better indicator of erythrocytosis than the hematocrit. Changes in the (A) hematocrit and (B) RBC count in a 37-year-old woman with PV-associated HVT treated with phlebotomy, Coumadin, and a transjugular intrahepatic portosystemic shunt (TIPS), demonstrating the correlation of the RBC but not the hematocrit with TIPS thrombosis (arrows) on 2 occasions. M/CU MM, millions per cubic millimeter; PCV, packed cell volume.
Patient evaluation
My evaluation begins with a medical history form, sent to patients in advance for completion at their leisure in a familiar environment (supplemental Exhibit A: Medical history form [available on the Blood Web site], available for noncommercial use). Created before the MPN patient symptom score,55 the form was designed to capture MPN-associated symptoms. An inquiry about pain with respect to location and intensity on an analog scale is conducted at the visit.
With respect to symptoms, visual disturbances, cognitive impairment, migraine, transient ischemic attacks (TIAs), aquagenic pruritus, erythromelalgia, and pica can be attributed directly to PV. Constitutional symptoms are unusual early in the disease except for fatigue, which has many progenitors56 : thyroid or cardiovascular disease, particularly in women; sleep apnea; depression; antidepressant drugs; MPN therapy (hydroxyurea, interferon, ruxolitinib); and pulmonary hypertension. Iron deficiency without anemia is not a cause of fatigue.57 Acid reflux may indicate a Helicobacter pylori infection, which is common in PV.58
On physical examination, I focus on the skin and mucous membranes for acne rosacea, plethora, glossitis, and cheilosis; the Darier sign to search for concomitant mastocytosis if there is a history of urticaria; sternal tenderness as a sign of disease activity; and liver and spleen size. Unless a patient is obese, I do not use ultrasound to detect or follow splenomegaly.
The initial laboratory evaluation includes a complete blood count and blood smear and a JAK2V617F assay. I do not obtain a marrow specimen for diagnostic purposes,37,51 or to screen for myelofibrosis in the absence of splenomegaly, a leukoerythroblastic reaction, unexplained anemia, or thrombocytopenia. I also obtain single-nucleotide polymorphism and next-generation sequencing assays under these circumstances. I always obtain a quantitative JAK2V617F VAF because MPN driver mutations are not mutually exclusive59 and the VAF permits assessment of disease burden and clonal dominance (Figure 2)60 ; it cannot be used prognostically12,61,62 but if >50%, ET is excluded.16 I do not obtain cytogenetics at diagnosis as these are usually uninformative.63 For thrombocytosis ≥900 000/mL, I obtain a ristocetin cofactor assay to look for acquired von Willebrand disease64 ; for leukocytosis, I obtain a uric acid level; and for the Darier sign, I obtain a serum tryptase level.
Risk assessment
Current therapeutic recommendations65 for chronic-phase PV stratify risk according to age and thrombosis history; phlebotomy and aspirin are recommended in patients <60 years of age. In patients >60 years of age, hydroxyurea for cytoreduction to prevent thrombosis is recommended, even though PV thrombosis is provoked and no study has proven that hydroxyurea prevents either arterial or venous thrombosis66-68 ; at best, it is a nitric oxide (NO) donor that inhibits platelet aggregation and prevents TIA uncontrolled by aspirin.66 The recommendations make no reference to control of splenomegaly, nor acknowledge that the impact of age on survival with PV is independent of the disease,69 or the very high risk of thrombosis in patients <60 years old.26 Furthermore, the recommendations are based on observational data70 involving patients with inadequate hematocrit control,71 and fail to specify that male and female patients have different target hematocrits.72 Recently, a genomic-based MPN prognostic calculator has been developed19 ; its ability to predict the risk of transformation during the chronic-phase PV remains to be established.
The recommendation to normalize blood counts in PV was not evidence based and had no impact on survival.73 No study, prospective, observational, or retrospective, has proved that leukocytosis74,75 or thrombocytosis76-78 causes thrombosis in PV. We also know that hydroxyurea-induced hematologic remission was not beneficial in terms of thrombosis prevention or survival but could be associated with myelofibrosis, massive splenomegaly, and leukemic transformation79 because hydroxyurea cannot eliminate the involved HSCs.80 In fact, neither antileukemic81,82 nor solid tumor therapy are capable of eradicating PV HSCs.
Patient 3
A 68-year-old man complained of occipital headaches. His levels and counts were as follows: hemoglobin level gram percentage, 18.9 g/dL RBCs, 6.8 × 1012/L; hematocrit, 0.54; MCV, 79 fL; ANC, 13 × 109/L; platelets, 676 × 109/L; and JAK2V617F VAF, 77%. Physical examination was normal. The patient was treated with phlebotomy. Four years after diagnosis, the patient was asymptomatic but his ANC was 34 × 109/L, his platelet count was 1 .1 × 109/L, his spleen extended 6 cm below the left costal margin, and his JAK2V617F VAF was 96%. Pegylated interferon (pegIFN) was started. The blood counts were reduced as was spleen size by 50%, though phlebotomies were still required. Subsequently, the patient developed rectal carcinoma and pegIFN was discontinued. The ANC was 15. 5 × 109/L and the platelets were 660 × 109/L. He received chemotherapy and radiotherapy before surgery and 3 cycles of chemotherapy afterward with a successful outcome. At that time, the ANC was 20. 9 × 109/L, the platelets were 888 × 109/L, and the spleen was palpable 3 cm below the left costal margin. The JAK2V617F VAF was 86%.
With respect to PPMF, neither the PMF International Prognostic Scoring System (IPSS)83 nor the Dynamic International Prognostic Scoring System (DIPSS)83,84 are useful for risk assessment, and a more specific risk-assessment score, the Myelofibrosis Secondary to PV and ET–Prognostic Model (MYSEC-PM), has been developed.35 Whether the newly developed MPN genomic scoring system19 will be equally useful in PPMF is unknown. What is important in PPMF is not the myelofibrosis but progressive splenomegaly often uncontrollable by chemotherapy or irradiation, which also poses management difficulties postsplenectomy due to thrombosis and extreme myeloproliferation.85
Treatment
Although I differ regarding the hematocrit target,11 I otherwise subscribe to Dameshek’s wisdom about PV therapy:
There is a tendency in medical practice—by no means limited to hematologists—to treat almost any condition as vigorously as possible. In hematology, this consists in attempting to change an abnormal number—whether this number is the hematocrit, white cell count or platelet count to get normal values, whether the patient needs it or not!86(p490)
For readers unfamiliar with PV history, there has always been prejudice against phlebotomy therapy.87 The Polycythemia Vera Study Group (PVSG) had 3 debates87 before including, at Dameshek’s insistence, a phlebotomy therapy–only control arm in PVSG-01, the first PV randomized controlled clinical trial (RCT)39 ; no PV RCT since has had a phlebotomy-only control arm.
PVSG-01 sought to determine whether chemotherapy or 32P supplemented by phlebotomy caused AML in therapy-naive patients; however, the trial was flawed because the initial hematocrit target, 52%, was changed to 45% 4 years after full enrollment; so phlebotomy-only patients were essentially untreated until then.88 Despite this, thrombosis frequency, though initially higher in the phlebotomy arm, was eventually equal in all 3 arms9 ; survival was superior in the phlebotomy-only arm, whereas the AML incidence was greater in the chemotherapy (13.5%) and 32P arms (10.2%) compared with phlebotomy only (1.5%).39 Furthermore, there was an increase in skin cancer and gastrointestinal tumors, lymphomas, and nonhematologic malignancies in the chemotherapy and 32P arms; observations confirming these results have since been continuously reported.40,74,89,90 These results notwithstanding, the PVSG adopted hydroxyurea, a nonalkylating agent, as the therapy of choice based on an observational study using the flawed PVSG-01 phlebotomy-only arm as a control.91
Hydroxyurea is similar to other drugs that impair DNA synthesis; they all cause tAML because they facilitate clonal expansion of HSC-bearing harmful mutations.92 Hydroxyurea is a skin tumor promoter93 that causes therapy-related AML (tAML) in association with 32P or alkylating agents94,95 or del17p42 and inhibits TP53 activation.96 Importantly, in a PV RCT with long-term follow-up, 24% of hydroxyurea-treated patients developed AML by 20 years.40
PV treatment has 2 goals: alleviating symptoms and prolonging survival through prevention of thrombosis, intractable splenomegaly, and leukemic transformation. Adequate phlebotomy therapy alleviates symptoms due to hyperviscosity but not severe migraine, aquagenic pruritus, or erythromelalgia. Control of thrombocytosis may be necessary for migraine relief or TIA, for which I prefer pegIFN to anagrelide; aquagenic pruritus has many therapeutic options depending on its severity: ruxolitinib, pegIFN, psoralen and ultraviolet A,97 and hydroxyurea. Erythromelalgia classically responds to aspirin.98 Thrombocytosis-induced von Willebrand syndrome usually does not cause spontaneous bleeding; for minor surgery or dental procedures, tranexamic acid or ε-aminocaproic acid suffice; for major surgery, platelet count reduction to achieve normal ristocetin cofactor activity is necessary.
Thrombosis is the most immediate threat to health in PV. Phlebotomy is the cornerstone of therapy; it completely addresses the Virchow triad by reducing the RCM and expanding the plasma volume.11 In newly diagnosed patients, this can be accomplished by daily or every-other-day procedures or all at once by erythrocytapheresis.99 The ultimate aim is to induce a state of chronic iron deficiency, which reduces phlebotomy frequency.
PV patients absorb iron maximally100 ; an increased phlebotomy requirement is a sign of excess body iron, not disease acceleration. Phlebotomy does not cause myelofibrosis,101 provoke thrombosis,102 or stimulate hematopoiesis because PV hematopoiesis is autonomous. Compliance is improved by giving patients a monthly phlebotomy appointment schedule for hematocrits ≥45% for men103 or ≥42% in women.72 To those arguing that the latter target needs formal proof, men have 10-fold higher testosterone levels than women and at every body weight, a woman’s normal RCM is ∼600 mL lower.104 Thus, at a hematocrit of 45%, a female PV patient has at least an excess of ∼600 mL of blood and much more with HVT,12 which frequently occurs with a normal hematocrit due to plasma volume expansion.32 Symptoms or thrombosis recurrence are the best evidence for inadequate phlebotomy therapy (Figure 4).
This is not a trivial issue because HVT is a unique and catastrophic feature of PV, primarily affecting young women105 (at my referral institution, the male-to-female ratio was 1:19; M. J. Walters and J.L.S., unpublished data); portal vein thrombosis is commoner in men. PV HVT emphasizes an additional hypercoagulable mechanism, NO scavenging, shared with sickle cell disease and paroxysmal nocturnal hemoglobinuria, in which HVT also occurs. In the latter 2, this is due to NO scavenging by plasma hemoglobin from intravascular hemolysis; in PV, it is due to NO scavenging by erythrocyte hemoglobin of the large RCM in the low flow rate splanchnic venous system because blood viscosity increases at low flow rates.106 As a consequence, phlebotomy therapy is mandatory in HVT, even when the presenting hematocrit is normal (Figure 4) because plasma volume expansion is always present masking the elevated RCM,12,32 and anticoagulation alone will be futile; I do not use chemotherapy in PV HVT.107
Pregnancy in PV is similar to HVT with respect to plasma volume expansion, but this is in part physiologic.108 In pregnancy, a phlebotomy hematocrit target of ≤45% essentially represents no treatment because the average normal pregnancy hematocrit is 33%.109 I maintain my pregnant PV patients at hematocrits <33%. This does not deprive the fetus of iron.110 I do not treat with aspirin or anticoagulation except in high-risk situations.111,112 If splenomegaly develops, pegIFN is safe during pregnancy.
Aspirin was recommended for primary thrombosis prevention in all PV patients without a contraindication, based on an RCT113 involving patients with hematocrits >45%; but this recommendation has been challenged because of the bleeding risk,114,115 especially in patients older than 60 years of age. I do not use aspirin in asymptomatic PV patients <50 years of age without cardiovascular risk factors unless they smoke or have diabetes, nor in patients ≥60 years of age because of the bleeding risk115-117 ; between the ages of 50 and 60 years, I follow the US Preventive Services Task Force (USPSTF) guidelines for prevention of cardiovascular disease.118
Targeted therapy
The discovery of JAK2 V617F and, as result, ruxolitinib, a JAK1/2 inhibitor,119 opened a new MPN therapeutic era. Together with pegIFN, which targets the involved HSCs (Figure 1A),120 we have 2 nonmyelotoxic, target-specific agents to treat PV. Ruxolitinib, unlike pegIFN, does not kill HSCs,121 making it unlikely to produce molecular remissions, and both drugs are not without significant side effects122,123 ; however, we are dealing with a neoplasm, and controlling splenomegaly and achieving molecular remissions while avoiding tAML are worthwhile goals.
Ruxolitinib proved to be effective in PPMF124 and chronic-phase PV125,126 ; provided durable symptom relief, blood count control, and reduction in splenomegaly; and was superior to hydroxyurea. We have no information, however, about its role in chronic-phase PV; in particular, its effectiveness compared with phlebotomy for hematocrit control and prevention of thrombosis, which insurers will want to know, as well as its long-term safety with respect to immunosuppression in this patient population.
We are also faced with the problem of how and when to use pegIFN; comparing it to hydroxyurea will not provide the answer because hydroxyurea does not affect the involved HSCs. Furthermore, as shown in Figure 5, most PV patients with an indolent phenotype can enjoy substantial longevity with phlebotomy alone but we also know that molecular remission is a 20% possibility.127,128
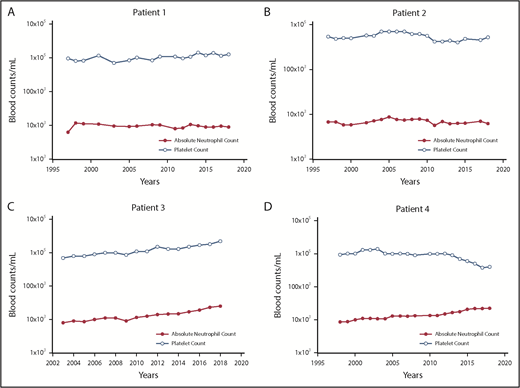
The changes in the platelet and neutrophil counts in 4 PV patients treated only with phlebotomy to maintain the hematocrit <45% in men and <42% in women. (A) Patient 1. A 57-year-old woman with PV for 39 years with a JAK2V617F VAF of 67%, who still requires periodic phlebotomies. (B) Patient 2. A 67-year-old woman with PV for 37 years with a JAK2V617F VAF of 50%, who still requires periodic phlebotomies. (C) Patient 3. A 77-year-old man with PV for 16 years with a JAK2V617F VAF of 67%, who does not require phlebotomy. (D) Patient 4. A 76-year-old woman with PV for 19 years with a JAK2V617F VAF of 77%, who does not require phlebotomy.
The changes in the platelet and neutrophil counts in 4 PV patients treated only with phlebotomy to maintain the hematocrit <45% in men and <42% in women. (A) Patient 1. A 57-year-old woman with PV for 39 years with a JAK2V617F VAF of 67%, who still requires periodic phlebotomies. (B) Patient 2. A 67-year-old woman with PV for 37 years with a JAK2V617F VAF of 50%, who still requires periodic phlebotomies. (C) Patient 3. A 77-year-old man with PV for 16 years with a JAK2V617F VAF of 67%, who does not require phlebotomy. (D) Patient 4. A 76-year-old woman with PV for 19 years with a JAK2V617F VAF of 77%, who does not require phlebotomy.
Patient 4
At 30 years of age, this patient had a hemoglobin gram percentage of 20 and a platelet count of 600 × 109/L associated with dizziness, headaches, and epistaxis. A diagnosis of PV was made and phlebotomy begun. At 39 years of age, she had not required phlebotomy for a year but had a severe migraine attack associated with a TIA. Her levels and counts were as follows: hemoglobin gram percentage, 11.5 g/dL; RBC, 5.7 × 1012/L; hematocrit, 0.37; MCV, 64 fL; ANC, 17.1 × 109/L; and platelets, 1.1 × 109/L. Her spleen was palpable at the left costal margin; the JAK2V617F VAF was 68%. Given the patient’s age and symptoms, she was started on 45 μg of pegIFN weekly. After a year, with dose escalation to 180 μg, she achieved hematologic remission, her spleen was nonpalpable, the JAK2V617F VAF was 1%, and pegIFN was discontinued. Two years later, she was clinically unchanged and the JAK2V617F VAF was 5.75%.
PegIFN can be also used to reduce established splenomegaly but not usually to normal size (patient 3); its role in PPMF is still under study.129 Therefore, because pegIFN is HSC-specific, in newly diagnosed PV patients, particularly age <60 years, I offer the option of pegIFN therapy; even if remission is not achieved, interferon effects appear to be durable (patient 3), as in chronic myeloid leukemia,130 and recent results in pegIFN-treated PV support this contention because patients who lost a hematologic response continued to obtain benefit with respect to symptoms.131 I start at 45 μg weekly and stay at this dose unless I do not obtain a significant effect. Because continuous pegIFN exposure can drive normal HSCs into cell cycle,132 and is associated with arrhythmias, hypothyroidism, and autoimmune events,123 I am not adverse to drug holidays. Because interferon stimulates erythropoiesis,120 phlebotomy often remains necessary.
Patient 5
A 52-year-old woman with PV for 23 years had progressive leukocytosis and splenomegaly. She was intolerant of recombinant interferon, thalidomide, hydroxyurea, and pegIFN, and developed thrombocytopenia from an experimental JAK2 inhibitor. She had fatigue, abdominal pain, easy satiety, pruritus, and diarrhea; she could not work. Her spleen was palpable 2 cm below the umbilicus. Her levels and counts were as follows: hemoglobin gram percentage, 11.0 g/dL; MCV, 70 fL; WBC, 35. 2 × 109/L with a left shift without blasts; and platelets, 31 × 109/L. The JAK2V617F VAF was 100%. The patient was given 15 mg of ruxolitinib daily. The platelet count fell to 7000/mL and platelet transfusions were required. The ruxolitinib dose was reduced and over 5 months the platelet count rose over 100 000/mL.133 Four years later, while on ruxolitinib, her hemoglobin gram percentage was 12.8 g/dL, her MCV was 89 fL, her WBC was 6.2 × 109/L, and her platelet count was 140 × 109/L. Her spleen was nonpalpable and she works full-time.
Ruxolitinib produces durable effects but fatigue, weight gain, immunosuppression, herpes zoster, opportunistic infections, skin cancer, and, rarely, lymphomas are drawbacks.122 Patients should receive pneumococcal and zoster vaccines.134,135 Taking a drug holiday due to toxicity does not preclude a response on rechallenge. In advanced PPMF with anemia, thrombocytopenia, and intractable splenomegaly, in addition to ruxolitinib or in place of it, I will use daily low-dose (50-100 mg) thalidomide and prednisone (40 mg daily with tapering over 1 month), which can improve blood counts and reduce circulating blasts and splenomegaly, though not uniformly.136 Combined therapy with ruxolitinib and azacytidine has proven useful in advanced PPMF137 not amenable to marrow transplantation; management of PV with leukemic transformation has been discussed recently in Blood.138
Conclusion
When an MPN is a diagnostic consideration, PV is the diagnosis of exclusion because PV is a hypercoagulable disorder. Thrombosis due to blood hyperviscosity is the immediate risk to health, and phlebotomy, not chemotherapy, to a sex-specific target hematocrit or red cell count, is the initial therapy of choice. PV is chronic illness with the potential for substantial longevity. The current therapeutic challenge is how to integrate the 2 available nonmyelotoxic target-specific drugs, ruxolitinib and pegIFN, to control PV symptoms and prevent the development of JAK2V617F-positive HSC clonal dominance and extramedullary hematopoiesis in those patients most at risk in accordance with the dictates of precision medicine.
The online version of this article contains a data supplement.
Acknowledgments
The author is grateful to his patients for continuously educating him about PV and making possible his clinical and basic research activities by their participation. The author is also grateful to his colleague Alison Moliterno whose research has informed his understanding of the MPN. The author apologizes to the many investigators whose significant publications could not be cited here because of space restrictions.
Authorship
Contribution: J.L.S. planned and wrote the manuscript.
Conflict-of-interest disclosure: The author declares no competing financial interests.
Correspondence: Jerry L. Spivak, School of Medicine, Johns Hopkins University, Traylor 924, 720 Rutland Ave, Baltimore, MD 21205; e-mail: jlspivak@jhmi.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal