Key Points
LSCs rely on cysteine for survival.
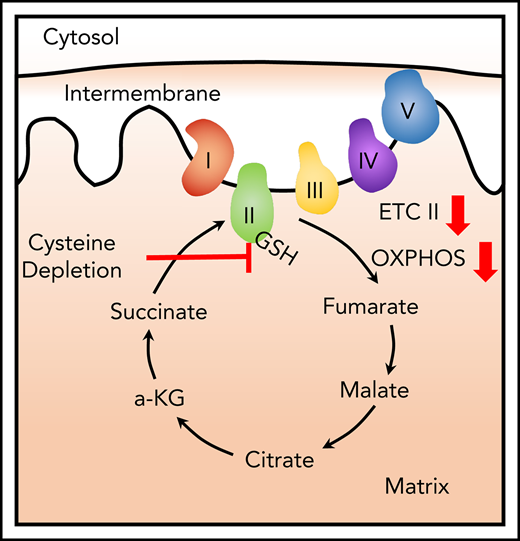
Cysteine metabolism is required for ETC II activity in LSCs.

Abstract
We have previously demonstrated that oxidative phosphorylation is required for the survival of human leukemia stem cells (LSCs) from patients with acute myeloid leukemia (AML). More recently, we demonstrated that LSCs in patients with de novo AML rely on amino acid metabolism to drive oxidative phosphorylation. Notably, although overall levels of amino acids contribute to LSC energy metabolism, our current findings suggest that cysteine may be of particular importance for LSC survival. We demonstrate that exogenous cysteine is metabolized exclusively to glutathione. Upon cysteine depletion, glutathione synthesis is impaired, leading to reduced glutathionylation of succinate dehydrogenase A (SDHA), a key component of electron transport chain complex (ETC) II. Loss of SDHA glutathionylation impairs ETC II activity, thereby inhibiting oxidative phosphorylation, reducing production of ATP, and leading to LSC death. Given the role of cysteine in driving LSC energy production, we tested cysteine depletion as a potential therapeutic strategy. Using a novel cysteine-degrading enzyme, we demonstrate selective eradication of LSCs, with no detectable effect on normal hematopoietic stem/progenitor cells. Together, these findings indicate that LSCs are aberrantly reliant on cysteine to sustain energy metabolism, and that targeting this axis may represent a useful therapeutic strategy.
Introduction
We have recently reported that amino acid metabolism is required for the survival of leukemia stem cells (LSCs) in patients with previously untreated acute myeloid leukemia (AML), and that inhibition of amino acid metabolism is a central component of the mechanism by which the highly effective regimen venetoclax + azacitidine works in this patient population.1,2 Detailed laboratory studies, combined with clinical observations of deep and durable remissions in many patients, indicate that venetoclax + azacitidine efficiently targets the LSC population in vivo.1,2
Notably, although our previous studies indicated that overall catabolism of amino acids into the trichloroacetic acid cycle is a key aspect of LSC biology, we do not yet understand how individual amino acids may be contributing. Thus, to ascertain whether there is a reliance of LSCs on specific amino acids, we have investigated how individual amino acids may be used to drive energy metabolism in LSCs. These studies indicate that cysteine is by far the single most important amino acid, and that catabolism of cysteine mediates synthesis of glutathione and posttranslational modification of succinate dehydrogenase A (SDHA), which in turn promotes oxidative phosphorylation and survival of LSCs.
Study design
Human specimens
AML specimens were obtained from apheresis product from patients with AML (supplemental Table 1, available on the Blood Web site) and mobilized peripheral blood or bone marrow from healthy donors who gave informed consent for sample procurement on the University of Colorado tissue procurement protocol. Specimens were cultured as previously described.1
Cell sorting
Primary AML specimens were stained with CD45 (BD, 571875), CD19 (BD, 555413), CD3 (BD, 557749), 4′,6-diamidino-2-phenylindole (EMD Millipore, 278298), and CellROX deep red (Thermo Fisher, C10422), and sorted using a BD FACSARIA. Cells with relatively low levels of reactive oxygen species (ROS-low LSCs) were identified as the cells with the 20% lowest ROS levels, and the ROS-high blasts were identified as the cells with the highest 20% ROS levels, as previously described.1,3
Global ultra-high pressure-liquid chromatography-mass spectrometry metabolomics
Metabolomics and metabolic flux using 13C3,15N-cysteine (Sigma-Aldrich, 658057) were performed via ultra-high pressure-liquid chromatography-mass spectrometry (Vanquish and Q Exactive, Thermo Fisher), as previously reported.4
Viability assays
Patient samples were sorted and cultured without amino acids or drugs for 24 hours. Viability was assessed by trypan blue (Gibco, 15250-071) staining and manual cell counting.
Normal HSC analysis
HSPCs from cord blood or bone marrow were cultured in indicated conditions for 24 hours, and CD34+ (BD, 572577), CD38+ (BD, 562288), and CD45+ (BD, 571875) percentages were quantified by flow cytometry (FACsCelesta, BD).
CFU assays
AML specimens or normal hematopoietic and progenitor cells (HSPCs) were cultured under indicated conditions for 24 hours before being plated in human methylcellulose (R&D systems HSC003). Colonies were counts 10 to 14 days after the initial plating.
Seahorse assays
XF96 (Agilent Technologies, 102417-100) extracellular flux assay kits were used to measure oxygen consumption, as previously described.1
Immunoprecipitation
Total cell lysates from cyst(e)inase-treated and glutathione-treated (Cayman Chemicals, 92614-59-0) cells collected and glutathionylation of SDHA was determined as previously described.1
ATP assay
ATP levels were quantified in cyst(e)inase-treated and glutathione-treated (Cayman Chemicals, 92614-59-0) cells according to the manufactures protocol (Roche, 11 699 709 001).
Electron transport chain complex II activity assay
AML cells were treated with cyst(e)inase or control inhibitors for 4 hours, samples were prepared, and enzyme activity was quantified according to the manufacturer’s protocol (Abcam, ab109908).
Results and discussion
To better understand the role of individual amino acids, we first interrogated the viability of primary AML cells after 24 hours of culture in which single amino acids were depleted. Three amino acids (cysteine, arginine, and glutamine) demonstrated a role in maintaining viability of bulk blast cells (supplemental Figure 1). However, only depletion of cysteine induced death of AML stem and progenitor cells (Figure 1A-B), defined as ROS-low LSCs, as previously described,1-3,5 and we therefore focused subsequent studies on cysteine. To investigate the role of cysteine in LSC biology, we first examined metabolism by incubating primary ROS-low LSCs with the stable isotope-labeled cysteine (13C315N) and measuring isotopologue distributions in cysteine metabolites by tracing the incorporation of 13C and 15N into the global metabolome of populations enriched for ROS-low LSCs.1,3 The heavy carbon and nitrogen atoms from cysteine were only detected in cysteine and glutathione, indicating that ROS-low LSCs can readily uptake cysteine and metabolize it to glutathione (Figure 1C). Notably, we detected no label in any other metabolites, indicating that ROS-low LSCs predominantly use cysteine to make glutathione.
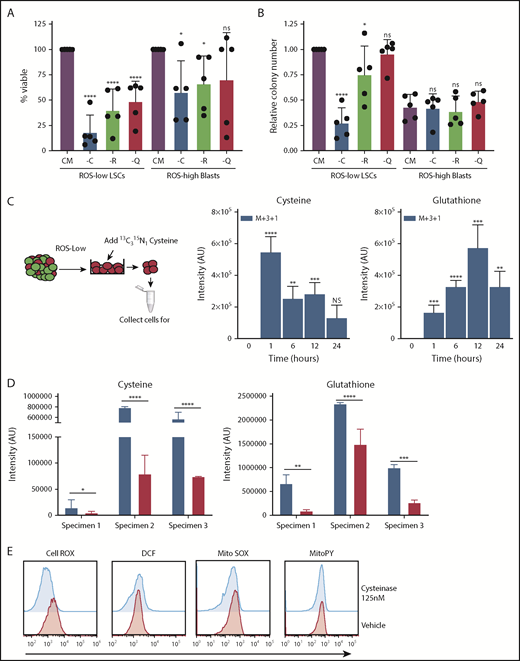
Cysteine depletion targets leukemia cells in a glutathione-dependent manner. (A) Viability of ROS-low LSCs and AML ROS-high blasts cultured without cysteine (-C), arginine (-R), or glutamine (-Q) for 24 hours normalized to cell cultured in complete media. (B) Colony number of primary AML ROS-low LSCs and AML ROS-high blasts after a 24-hour culture with (CM) or without cysteine (-C), arginine (-R), or glutamine (-Q). The methylcellulose contained all amino acids. Data were normalized to colony number in CM ROS-low LSCs. (C) Stable isotope-labeled cysteine and glutathione in ROS-low LSCs after incubation with cysteine (13C315N) for 1, 6, 12, or 24 hours. Y-axis represents intensity of metabolite detected by mass spectroscopy in arbitrary units (AU). The experiment was run in replicates of 5. (D) Cysteine and glutathione levels in ROS-low LSCs 8 hours postincubation with 125 nM cyst(e)inase. (E) Representative data of levels of cellular (measured by CellROX and DCF) and mitochondrial (measured by MitoSOX and MitoPY) reactive oxygen species in vehicle and cyst(e)inase-treated cells for 8 hours. Statistical significance was determined by a 2-way analysis of variance analysis. *P < .05; **P < .01; ***P < .005; ****P < .001.
Cysteine depletion targets leukemia cells in a glutathione-dependent manner. (A) Viability of ROS-low LSCs and AML ROS-high blasts cultured without cysteine (-C), arginine (-R), or glutamine (-Q) for 24 hours normalized to cell cultured in complete media. (B) Colony number of primary AML ROS-low LSCs and AML ROS-high blasts after a 24-hour culture with (CM) or without cysteine (-C), arginine (-R), or glutamine (-Q). The methylcellulose contained all amino acids. Data were normalized to colony number in CM ROS-low LSCs. (C) Stable isotope-labeled cysteine and glutathione in ROS-low LSCs after incubation with cysteine (13C315N) for 1, 6, 12, or 24 hours. Y-axis represents intensity of metabolite detected by mass spectroscopy in arbitrary units (AU). The experiment was run in replicates of 5. (D) Cysteine and glutathione levels in ROS-low LSCs 8 hours postincubation with 125 nM cyst(e)inase. (E) Representative data of levels of cellular (measured by CellROX and DCF) and mitochondrial (measured by MitoSOX and MitoPY) reactive oxygen species in vehicle and cyst(e)inase-treated cells for 8 hours. Statistical significance was determined by a 2-way analysis of variance analysis. *P < .05; **P < .01; ***P < .005; ****P < .001.
To transition our studies to a context that may have more translational relevance, we performed cysteine depletion using an engineered human cysteine-degrading enzyme, cyst(e)inase.6 As shown in Figure 1D, culture of primary ROS-low LSCs with cyst(e)inase strongly reduces intracellular glutathione levels and other glutathione-related metabolites (Figure 1D; supplemental Figure 2A). Further, we confirmed that cysteine depletion decreased glutathione levels in ROS-low LSCs by culturing ROS-low LSCs in media lacking cysteine (supplemental Figure 2B). Given the prevalent role of glutathione in regulating cellular oxidative state (via management of ROS), we expected treatment with cyst(e)inase to increase ROS.7 Surprisingly, analysis with multiple ROS probes show no consistent detectable change in oxidative state after cyst(e)inase treatment (Figure 1E; supplemental Figure 3A). Further, we interrogated changes in expression of HO1 and CHOP, genes known to be induced by ROS in LSCs. We also tested the ability of N-acetylcysteine to rescue the decreased viability observed on cyst(e)inase treatment. We did not observe any significant changes in ROS-induced gene expression or changes in viability on N-acetylcysteine pretreatment (supplemental Figure 3). Because the decrease in glutathione levels is not absolute, as previous studies in LSCs have shown,8 we hypothesized that the decreased level of glutathione may not be enough to elicit a change in ROS.
Given the lack of evidence for glutathione’s role in ROS management, we next examined alternate pathways known to be regulated by glutathione, focusing on protein glutathionylation. To this end, we recently reported that treatment of AML cells with venetoclax + azacitidine inhibits glutathionylation of SDHA,1 an important regulator of electron transport complex II (ETC II) activity.9 Notably, previous studies by Cole et al have demonstrated that ETC II activity is essential for LSC survival.10 Thus, we hypothesized that reduced cysteine uptake mediated by venetoclax + azacitidine1 treatment might be the specific mechanism underlying inhibition of ETC II. Supporting this hypothesis, cyst(e)inase treatment resulted in an accumulation of succinate the substrate for ETC II (supplemental Figure 4A) and no other changes in mitochondrial metabolites mediated by glutathionylated proteins such as isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, or enzymes involved in fatty acid oxidation (supplemental Figure 4B). Furthermore, ROS-low LSCs were insensitive to inhibition of another glutathionylated mitochondrial protein, ETC I (supplemental Figure 4C), suggesting that ETC I function is not essential in LSC survival in the context of glutathione loss. Overall, these data suggest ETC II is the promising enzyme to further pursue, but does not prove that other enzymes involved in OXPHOS are unchanged by cysteine depletion.
Consistent with the metabolic data upon cysteine depletion with cyst(e)inase, we observed a marked reduction of ETC II activity, as well as a consequent reduction in ATP (Figure 2A-B). Further, cyst(e)inase treatment decreased oxidative phosphorylation and mitochondrial ATP production (supplemental Figure 5). Direct analysis of SDHA by immunoblot (Figure 2C; supplemental Figure 6) confirms that cyst(e)inase treatment reduces glutathionylation. Importantly, for all the findings in Figure 2A-C, preincubation of cells with a cell-permeable form of glutathione completely rescues the effects of cyst(e)inase treatment on ETC activity. Together, these data demonstrate that cysteine depletion inhibits production of glutathione, which impairs glutathionylation of SDHA. The consequence of these biochemical events is impaired ETC activity, which reduces production of ATP.
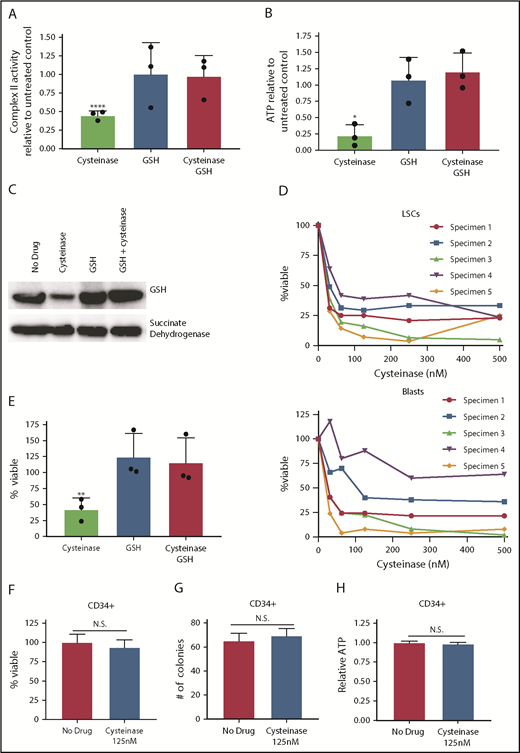
Glutathione is essential for energy metabolism in AML. (A) Complex II activity upon a 4-hour 125 nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. Data normalized to no treatment control. (B) ATP levels on a 4-hour 125-nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. Data normalized to no treatment control. (C) Western blot showing glutathione and succinate dehydrogenase A levels from succinate dehydrogenase A immunoprecipitation on a 4-hour 125-nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. (D) Cell viability of ROS-low LSCs or ROS-high AML blasts pon increasing concentrations of cysteine depleting enzyme, cyst(e)inase. AML specimens 1 and 3 are sensitive to venetoclax with azacitidine, and AML specimens 2, 4, and 5 are resistant to venetoclax with azacitidine.2 (E) Viability of AML cells after a 24-hour 125-nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. Data normalized to no treatment control. (F) Viability of CD34+ cells isolated from cord blood samples after a 24-hour 125 nM cyst(e)inase treatment. (G) Colony-forming number from CD34+ cells isolated from cord blood samples after a 24-hour, 125-nM cyst(e)inase treatment. (H) ATP level CD34+ cells isolated from cord blood samples after a 4-hour, 125-nM cyst(e)inase treatment. Statistical significance was determined by a 2-way analysis of variance analysis or Student’s t-test, where appropriate. *P < .05; **P < .01; ****P < .001.
Glutathione is essential for energy metabolism in AML. (A) Complex II activity upon a 4-hour 125 nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. Data normalized to no treatment control. (B) ATP levels on a 4-hour 125-nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. Data normalized to no treatment control. (C) Western blot showing glutathione and succinate dehydrogenase A levels from succinate dehydrogenase A immunoprecipitation on a 4-hour 125-nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. (D) Cell viability of ROS-low LSCs or ROS-high AML blasts pon increasing concentrations of cysteine depleting enzyme, cyst(e)inase. AML specimens 1 and 3 are sensitive to venetoclax with azacitidine, and AML specimens 2, 4, and 5 are resistant to venetoclax with azacitidine.2 (E) Viability of AML cells after a 24-hour 125-nM cyst(e)inase treatment with or without a 2-hour pretreatment with 100 µM cell-permeable glutathione. Data normalized to no treatment control. (F) Viability of CD34+ cells isolated from cord blood samples after a 24-hour 125 nM cyst(e)inase treatment. (G) Colony-forming number from CD34+ cells isolated from cord blood samples after a 24-hour, 125-nM cyst(e)inase treatment. (H) ATP level CD34+ cells isolated from cord blood samples after a 4-hour, 125-nM cyst(e)inase treatment. Statistical significance was determined by a 2-way analysis of variance analysis or Student’s t-test, where appropriate. *P < .05; **P < .01; ****P < .001.
Last, we sought to assess the biological selectivity of cysteine depletion in AML vs normal cell types. We observed that cell viability was reduced in both ROS-low LSCs and AML ROS-high blasts on treatment with cyst(e)inase (Figure 2D). Analogous to the data in Figures 2A-C, preincubation of AML cells with cell-permeable glutathione completely rescues the cytotoxicity of cyst(e)inase (Figure 2E). Importantly, AML specimens from patients with both de novo and relapsed/refractory AML were used for this analysis (supplemental Table 1). Previous clinical studies have indicated that relapsed/refractory LSCs are less sensitive to venetoclax with azacitidine treatment.11 In addition, we have previously shown these same relapsed/refractory specimens to be resistant to venetoclax with azacitidine,2 suggesting that cysteine depletion may provide a strategy to target ven/aza resistant LSCs. To determine why cysteine depletion could effectively target relapsed/refractory LSCs and venetoclax with azacitidine did not, we measured cysteine and glutathione levels on venetoclax and azacitidine treatment in venetoclax with azacitidine-resistant LSCs. Venetoclax with azacitidine treatment decreased cysteine and glutathione in venetoclax with azacitidine sensitive, as we have previously reported,1,2 but not in resistant LSCs (supplemental Figure 7A). Furthermore, combination therapy and not single-agent venetoclax or azacitidine decreased cysteine levels significantly in sensitivity LSCs (supplemental Figure 7B). These data suggest that loss of venetoclax with azacitidine sensitivity may be a result of lack of inhibiting these essential metabolic targets and not loss of dependency on glutathione metabolism. Importantly, neither cyst(e)inase treatment nor cysteine media depletion affects the viability or colony-forming potential of normal human CD34+ cells (Figure 2F-G; supplemental Figure 8). As expected, given the lack of toxicity, cyst(e)inase treatment also did not reduce ATP levels in normal CD34+ cells (Figure 2H). These data indicate that ROS-low LSCs are selectively dependent relative to normal HSPCs on cysteine to maintain energy metabolism.
In conclusion, we have shown that primitive AML cells require exogenous cysteine to drive synthesis of glutathione, which is important for modification of SDHA and activity of ETC II. Inhibition of cysteine metabolism alone is sufficient to substantially impair energy metabolism in ROS-low LSCs, but not HSPCs cells. In addition, as we have previously reported, ROS-low LSCs metabolize all amino acids into the trichloroacetic acid cycle as a means to fuel oxidative phosphorylation, which is targeted by venetoclax + azacitidine.1 Our new findings reveal that cysteine, as an individual amino acid, is essential for ETC activity. The consequence is impaired oxidative phosphorylation and selective targeting of ROS-low LSCs. Clinically, targeting cysteine metabolism in ROS-low LSCs may be accomplished by therapies designed to specifically degrade cysteine6 or by venetoclax + azacitidine, which we have shown decreases global amino acid levels.1 In addition, our current studies have helped to elucidate essential components of the venetoclax with azacitidine mechanism; namely, decreased cysteine levels, glutathione levels, and oxidative phosphorylation. In addition, our data suggest that induction of ROS by venetoclax with azacitidine may not be playing a functional role in cell death. Furthermore, as suggested by data shown in supplemental Figure 9, cysteine levels may serve as a biomarker of venetoclax response.
For original data, please contact Craig Jordan at craig.jordan@ucdenver.edu
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Giulia Agnello and Scott Rowlinson from Aeglea Biotherapeutics for providing the cyst(e)inase compound. In addition, the authors thank the University of Colorado Hematology Clinical Trials Unit for help in acquisition of patient samples. The authors also acknowledge the Molecular and Cellular Analytical Core within the Colorado Nutrition and Obesity Research Center for the use of the Seahorse Analyzer.
This work was supported by the American Cancer Society 25A5072, the Colorado Clinical and Translational Sciences Institute, and the Cancer League of Colorado (C.L.J.); the V Foundation (J.D.); National Institutes of Health, National Cancer Institute grant R01 5 R01 CA200707 (C.T.J); and a Leukemia and Lymphoma Society Specialized Center of Research grant (principal investigator, C.T.J.). C.T.J. is supported by the Nancy Carroll Allen Endowed Chair.
Authorship
Contribution: C.L.J. and B.M.S. designed and performed the research; collected, analyzed, and interpreted the data; performed the statistical analysis; and wrote the manuscript. A.D., R.C.-H., and J.A.R. performed metabolomics experiments; collected, analyzed, and interpreted metabolomics data; and wrote the manuscript. S.P., A.G., and N.K., performed experiments. J.D. and D.A.P. directed the experiments and wrote the manuscript. C.T.J. designed and directed the research, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: D.A.P. receives research funding from and has served as a consultant for AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Craig T. Jordan, University of Colorado-Anschutz Medical Campus, 12700 E 19th Ave, Rm 10016, Aurora, CO 80045; e-mail: craig.jordan@ucdenver.edu.