Key Points
Clinically available miR-29b exerts potent antileukemic activity in primary CLL cells in vitro and in an hROR1 × TCL1 mouse model in vivo.
Leukemia-targeted delivery of miR-29b via ROR1 induces epigenetic reprogramming and p21-dependent cell cycle arrest in vivo.

Abstract
Chronic lymphocytic leukemia (CLL) occurs in 2 major forms: aggressive and indolent. Low miR-29b expression in aggressive CLL is associated with poor prognosis. Indiscriminate miR-29b overexpression in the B-lineage of mice causes aberrance, thus warranting the need for selective introduction of miR-29b into B-CLL cells for therapeutic benefit. The oncofetal antigen receptor tyrosine kinase orphan receptor 1 (ROR1) is expressed on malignant B-CLL cells, but not normal B cells, encouraging us with ROR1-targeted delivery for therapeutic miRs. Here, we describe targeted delivery of miR-29b to ROR1+ CLL cells leading to downregulation of DNMT1 and DNMT3A, modulation of global DNA methylation, decreased SP1, and increased p21 expression in cell lines and primary CLL cells in vitro. Furthermore, using an Eμ-TCL1 mouse model expressing human ROR1, we report the therapeutic benefit of enhanced survival via cellular reprograming by downregulation of DNMT1 and DNMT3A in vivo. Gene expression profiling of engrafted murine leukemia identified reprogramming of cell cycle regulators with decreased SP1 and increased p21 expression after targeted miR-29b treatment. This finding was confirmed by protein modulation, leading to cell cycle arrest and survival benefit in vivo. Importantly, SP1 knockdown results in p21-dependent compensation of the miR-29b effect on cell cycle arrest. These studies form a basis for leukemic cell–targeted delivery of miR-29b as a promising therapeutic approach for CLL and other ROR1+ B-cell malignancies.
Introduction
Despite the extensive elucidation of the role of noncoding RNAs, including microRNAs (miRs) in health and disease, the transition of miRs as therapeutic agents to clinical applications has been limited.1 Part of this difficulty is ascribed to the pleiotropic effects of these noncoding RNAs on multiple targets and differential roles in multiple cell types. Attempts to study the therapeutic evaluation of miRs in disease models have been restricted to human cell lines in immunocompromised xenograft models that preclude influence of the immune compartment. For example, miR-34 mimic (MRX34; Mirna Therapeutics) entered a multicenter phase 1 trial of solid tumors in 2013 with significant tumor regression but was terminated due to the immune-related serious adverse events.2 The immune deficiency in the preclinical models of miR therapy evaluation does not reflect the clinical scenario and precludes investigating the effect of the miRs on the immune microenvironment, thus limiting the development of miR-based therapy for clinical application.
Noncoding RNA profiling in aggressive CLL revealed lower expression of miR-29b that is associated with reduced survival, increased drug resistance, and poor prognosis via upregulation of antiapoptotic proteins such as myeloid leukemia cell differentiation protein 1 (MCL1) and oncogenic T-cell leukemia protein 1 (TCL1).3 Although increasing miR-29b levels in aggressive CLL is therefore an attractive approach, the pleiotropic effects of miR-29b in physiological homeostasis pose a challenge. For example, treatment with miR-29b in muscle cells in vivo promotes atrophy via reduction of its targeting of insulin-like growth factor 1 and phosphatidylinositol 3-kinase (p85α).4 Homeostasis of miR-29b and regulation of its targets, such as BH3-only BCL2 family proteins, are critical during neuronal maturation and apoptosis.5 Sustained overexpression of miR-29b starting as early as the B-cell precursor stage in Eμ-miR-29b transgenic mice leads to the development of a CD5+CD19+ B-cell population, highlighting the need for fine modulation of miR-29b expression in normal B-cell development.6 These observations warrant selective introduction of miR-29b into mature leukemic CLL cells, thus sparing normal precursor and mature B cells and obviating adverse effects on nonintended target cells as a potential therapeutic modality for patients with aggressive CLL.7 In addition, short circulation half-life and limited cellular uptake pose a challenge for miR-based therapies, as has been the case for other nucleic acid–based agents.
To overcome these limitations and to facilitate selective and efficient delivery of miR-29b to CLL cells, we developed an immuno-nanoparticle–based miR-29b delivery formulation with selectivity to CLL cells but not normal B cells via targeting them to receptor tyrosine kinase orphan receptor 1 (ROR1), which is expressed in >95% of CLL cells but not normal B cells.8 We show that ROR1-targeted nanoparticles mediate strong internalization and uptake of encapsulated miR-29b, resulting in epigenetic reprogramming by downregulating DNMT1 and DNMT3A.9,10 Using a novel human ROR1-expressing murine model of CLL, we describe the therapeutic benefit of delivering miR-29b to ROR1-expressing leukemic cells in vivo. This strategy resulted in downregulation of DNMTs, reduction of DNA methylation and SP1, p21-dependent cell cycle deregulation, and antileukemic activity.
Materials and methods
Nanoparticle preparation
Synthetic 2′-MeOPSmiR-29b and 2′-MeOPS scrambled miR (2′-MeOPSmiR-sc) oligonucleotides were purchased from Girindus. The sequences of these oligos were the same as previously reported.11 The lipid components were 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 1,2-dimyristoyl-sn-glycero-methoxypolyethylene glycol (molecular weight, ∼2000; Avanti Polar Lipids), and linoleic acid (MilliporeSigma). The molar ratio of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine/linoleic acid/1,2-dimyristoyl-sn-glycero-methoxypolyethylene glycol was 50:48:2. Synthetic 2′-MeOPSmiR were mixed with polyethylenimine (molecular weight, ∼2000; MilliporeSigma) at room temperature. The N/P ratio (the ratio of moles of the amine group of polyethylenimine to those of the phosphate groups of DNA) was 10:1. The liposome nanoparticles were generated by injecting the lipid complex into 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer (pH 7.4). The mass ratio of lipid complex to miRs was 10:1. Lipid complex nanoparticles containing the miR were vortexed and sonicated to avoid aggregation. A postinsertion method was adopted to incorporate 2A2-antibody ligand onto the miR-loaded nanoparticle as previously described.12,13 The immunoglobulin G (IgG) control nanoparticle followed the same systematic procedure with mouse IgG1κ (Thermo Fisher Scientific). The characterization of the immuno-nanoparticle and its cargos are described in supplemental Table 1 (available on the Blood Web site).
Cells
Peripheral blood was obtained from patients with CLL after receipt of written informed consent under an institutional review board–approved protocol. CLL cells were isolated by using Ficoll density gradient centrifugation (Ficoll-Paque Plus; Amersham Biosciences) according to the manufacturer’s instructions. Normal B cells were collected from Leukopaks from the American Red Cross of Central Ohio. Cells were cultured in RPMI 1640 media (Thermo Fisher Scientific) with 10% heat-inactivated fetal bovine serum (MilliporeSigma); 2 mM L-glutamine and penicillin (100 U/mL); and streptomycin (100 μg/mL) (Thermo Fisher Scientific) at 37°C with 5% carbon dioxide.
Flow cytometry
The internalization assay was performed by measuring intracellular fluorescence-labeled nanoparticles within cells. Treated cells were washed with ice-cold high-glycine stripping buffer (100 mM glycine, 100 mM NaCl, pH 2.5) for 2 minutes to remove the surface-bound nanoparticles. Cell death was measured by using Near-IR staining with flow cytometry (Thermo Fisher Scientific). Approximately 1 million cells in 100 μL of fetal calf serum buffer (Thermo Fisher Scientific) were stained with antibody cocktails for 15 minutes per the manufacturer’s instruction. Events were captured on a Gallios Flow Cytometer and analyzed by using Kaluza (Beckman Coulter) and FCS Express (De Novo Software) software.
Imaging by confocal microscopy
Primary CLL cells were incubated with Cy5-nucleotide-labeled nanoparticles for 16 hours and fixed in 4% paraformaldehyde. Fixed cells were stained with goat polyclonal anti-ROR1 antibody (R&D Systems) and Hoechst 33258 stain (MilliporeSigma) for nuclei. Stained cells were mounted on poly-D-lysine–coated glass slides (MilliporeSigma) and examined with an Olympus FV1000 confocal microscope (Olympus). Image capture and analysis were performed by using ImageJ software (https://imagej.nih.gov/ij/).
Immunoblot analysis
Cell lysates were prepared and quantified by using the bicinchoninic acid method (Thermo Fisher Scientific). Lysates were separated by using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to 0.2-μm nitrocellulose membranes (Bio-Rad). Blots were probed with indicated primary antibodies followed by horseradish peroxidase–conjugated goat anti-rabbit or goat anti-mouse IgG (Bio-Rad). Anti-DNMT1, SP1, DNMT3A, BCl-2, MCL1, and actin antibodies were purchased from Santa Cruz Biotechnology. The intensity of the resulting bands was analyzed by using ImageJ software.
RNA extraction and analysis
Total RNA was extracted by using TRIzol (Thermo Fisher Scientific), and complementary DNA was prepared by using random hexamer priming. Real-time polymerase chain reaction was performed by using TaqMan (Thermo Fisher Scientific) gene expression assay primer/probe sets for DNMT1, DNMT3A, and SP1. The signal was measured on a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). The expression of target genes relative to the internal control gene was calculated by using the threshold cycle number (Ct). The relative target gene expression for each condition was normalized to vehicle control and fold change determined by using the comparative method (2ΔΔCt). A hybridization-based enzyme-linked immunosorbent assay was used to quantify 2′-MeOPSmiR-29b concentration in cell pellets and mouse organs, as previously reported.11 Sequence analysis of RNA (RNA-seq) library generation and data analysis were performed at The Ohio State University Comprehensive Cancer Center Genomics Shared Resource (see methods section of supplemental Information).
Global DNA methylation analysis
Genomic DNA was extracted from cells by using the DNeasy Tissue Kit (Qiagen), hydrolyzed, and analyzed with liquid chromatography–tandem mass spectrometry as previously reported.14 The ratio of 5-methyl-2′-deoxycytidine to the internal standard 2-deoxyguanosine in mass signal was used to quantify the global DNA methylation (GDM) level.
Engraftment of hROR1 × TCL1 mice
All animal experiments were conducted under protocols approved by the OSU Institutional Animal Care and Use Committee. Eμ-ROR1 transgenic mice were crossed with Eμ-TCL1, a well-established mouse model of CLL, to generate Eμ-ROR1-TCL1 double transgenic animals as previously described.15 Live leukemic splenocytes (107 cells) from diseased hROR1 × TCL1 mice were collected and serially engrafted into 6- to 8-week-old C57BL/6 wild-type recipient mice via the tail vein. The recipient mice developed reproducible CD19+CD5+hROR1–expressing leukemic cells 2 to 3 weeks postengraftment. Upon achieving the peripheral CD19+CD5+hROR1+ leukemic population to 5% of total white blood cells, mice were treated with phosphate-buffered saline, 2A2-miR-29b-ILP, IgG-miR-29b-ILP, or 2A2-scramble-ILP (1.5 mg/kg per day, 3 times per week).
Statistical analysis
All statistical analyses were performed in the OSU Center for Biostatistics. For experiments using samples from the same patients, mixed effect models were used to take the dependency of these observations into consideration. For experiments involving multiple independent groups, general linear models were used. Contrasts in models that answer primary questions directly were tested. For the mouse survival study, the log-rank test was used to test the difference between the 2 survival curves. The Holm procedure was applied to adjust for multiplicity. The overall family-wise type I error rate was controlled at α = 0.05. SAS version 9.3 was used for all statistical analyses (SAS Institute, Inc).
Results
Characterization of anti-ROR1 antibody immunoliposome (2A2-ILP) for miR-29b delivery
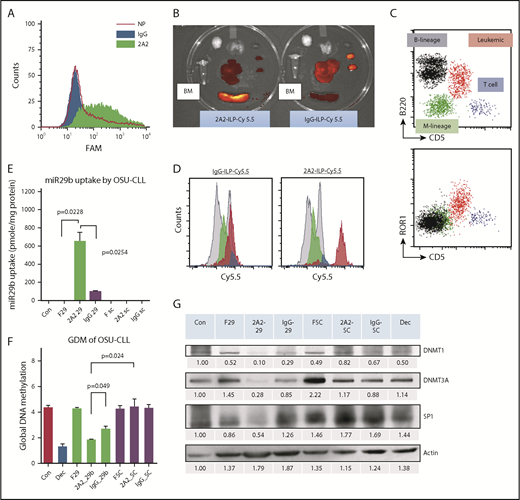
To selectively target miR-29b to leukemic CLL B cells, we developed a liposomal nanoparticle target ROR1, which is a surface antigen expressed on primary CLL cells but not on normal B cells. Clone 2A2, an anti-human ROR1 antibody-encapsulated miR immune-liposomal formulation (ILP) of uniform size (mean diameter, 112.8 ± 0.7 nm) and zeta potential (3.7 ± 1.0 mV), was efficiently internalized. The specific uptake of 2A2-ILP over control IgG-ILP by primary CLL cells was optimized by using varying ratios of antibody and lipid complex formulation (Figure 1A; supplemental Figure 1). To evaluate the delivery efficacy of ROR1-targeted 2A2-ILP in vivo, we developed a human ROR1-bearing CD5+CD19+ murine CLL leukemic mouse model.15 Although the human ROR1-directed 2A2-Cy5.5-miR-ILP targeted to human ROR1-expressing leukemia cells enriched in spleen, the control IgG-Cy5.5-miR-ILP accumulated predominantly in liver and kidneys 24 hours postinjection, as shown with the use of an in vivo imaging system (IVIS; PerkinElmer) (Figure 1B). To further identify the specific targeting of 2A2-ILP in bone marrow, we flushed out the bone marrow mononuclear cells (BMMC) and identified populations of myeloid (green), normal B-lymphoid (black), T-lymphoid (blue), and hROR1-expressing CD5+CD19+ leukemic (red) cells by using flow cytometry (Figure 1C). With the mean fluorescence intensity evidence, the 2A2-Cy5.5-miR-ILP accumulated in human ROR1-expressing leukemic cells in BMMC, whereas the control IgG-Cy5.5-miR-ILP had much less specificity (Figure 1D).
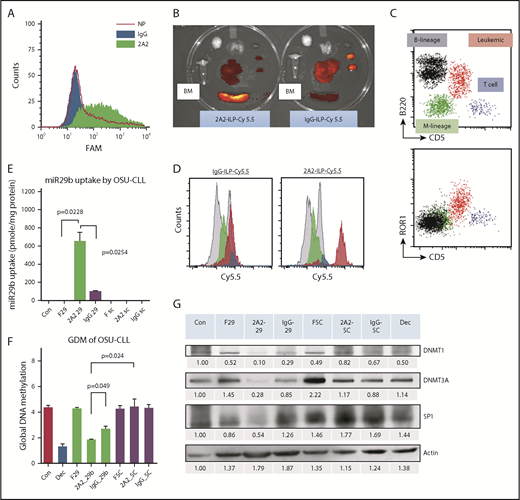
Characterization of selectivity of 2A2-ILP targeting to surface ROR1 on primary CLL cells. (A) The specific uptake of 2A2-ILP over control IgG-ILP by primary CLL was measure by intensity of fluorescence. (B) The in vivo selectivity of 2A2-ILP over IgG-ILP in organs from mice engrafted with hROR1 × TCL1 splenocytes. (C) The invasion of CD5+B220+ROR1+ leukemic cells (red) in BMMC of hROR1 × TCL1 mouse model. (D) The in vivo selectivity of 2A2-ILP over IgG-ILP recognizing ROR1+ leukemic cells in BMMC from mice engrafted with hROR1 × TCL1 splenocytes. (E) Treatment with miR-29b–encapsulated 2A2-ILP for 24 hours increased levels of intracellular miR-29b ∼600-fold over free form and sevenfold over isotype control IgG-ILP delivery in OSU-CLL cell line. (F29, free-form of miR-29b; SC, scrambled miR.) (F) Two-week treatment with 2A2-miR-29b-ILP significantly reduced the GDM of the CLL cell line by 58% compared with scramble in 2A2-ILP control. (G) Selective delivery of miR-29b decreases DNMTs and SP1 in OSU-CLL cell lines after 48 hours. Decitabine (Dec, 100 nM) was used as a positive control of hypomethylating agent.
Characterization of selectivity of 2A2-ILP targeting to surface ROR1 on primary CLL cells. (A) The specific uptake of 2A2-ILP over control IgG-ILP by primary CLL was measure by intensity of fluorescence. (B) The in vivo selectivity of 2A2-ILP over IgG-ILP in organs from mice engrafted with hROR1 × TCL1 splenocytes. (C) The invasion of CD5+B220+ROR1+ leukemic cells (red) in BMMC of hROR1 × TCL1 mouse model. (D) The in vivo selectivity of 2A2-ILP over IgG-ILP recognizing ROR1+ leukemic cells in BMMC from mice engrafted with hROR1 × TCL1 splenocytes. (E) Treatment with miR-29b–encapsulated 2A2-ILP for 24 hours increased levels of intracellular miR-29b ∼600-fold over free form and sevenfold over isotype control IgG-ILP delivery in OSU-CLL cell line. (F29, free-form of miR-29b; SC, scrambled miR.) (F) Two-week treatment with 2A2-miR-29b-ILP significantly reduced the GDM of the CLL cell line by 58% compared with scramble in 2A2-ILP control. (G) Selective delivery of miR-29b decreases DNMTs and SP1 in OSU-CLL cell lines after 48 hours. Decitabine (Dec, 100 nM) was used as a positive control of hypomethylating agent.
To validate the uptake and modulation of previously reported targets of miR-29b, we used OSU-CLL,16 a CLL patient–derived cell line with surface expression of ROR1 antigen (supplemental Figure 2). Treatment with miR-29b–encapsulated 2A2-ILP (2A2-miR-29b-ILP) increased levels of intracellular miR-29b ∼600-fold over the free form and sevenfold over isotype control IgG-ILP delivery (P = .0254; n = 3) (Figure 1E). The long-term effect of suppression of DNMTs was measured via the GDM of the OSU-CLL cells after 2 weeks treatment according to mass spectrometry as previously described.14 Treatment with 2A2-miR-29b-ILP significantly reduced GDM by 58% compared with 2A2-scramble-ILP control (2A2-miR-29b-ILP, 1.83 ± 0.06%; 2A2-scramble-ILP, 4.42 ± 0.35; P = .024; n = 3) (Figure 1F). Consistent with previous reports with miR-29b,9,10 we observed downregulation of DNMT1, DNMT3A, and SP1 in OSU-CLL cells incubated with 2A2-miR-29b-ILP but not with 2A2-scramble-ILP control or less in control miR-29b with IgG-ILP (Figure 1G; supplemental Figure 3A-C). In addition, the long-term effect of 2A2-miR-29b-ILP reduced the cell viability (supplemental Figure 3D) and growth (supplemental Figure 3E) in OSU-CLL cells.
Antileukemic activity of 2A2-miR-29b-ILP in primary CLL cells
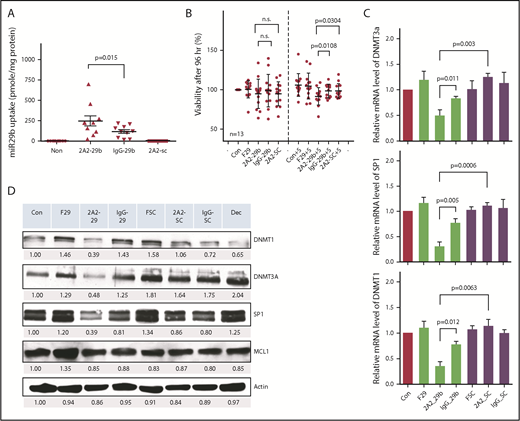
We further validated the antileukemic activity of 2A2-miR-29b-ILP with primary CLL cells from patients. Treatment of ROR1+ primary CLL cells with 2A2-miR-29b-ILP for 24 hours exhibited twofold higher efficiency of miR-29b uptake compared with IgG-miR-29b-ILP control (2A2-miR-29b-ILP, 271.6 ± 64.7; IgG-miR-29b-ILP, 127.8 ± 22.7; P = .015; n = 9) (Figure 2A). Although we observed no significant effect of miR-29b on the viability of primary CLL cells after 48 hours (supplemental Figure 4A-B), the long-term treatment of 2A2-miR-29b-ILP suppressed cell viability by ∼10% after 96 hours under the coculture of HS-5 stromal cells (percentage of viability: 2A2-29b-S, 91.6 ± 10.9; IgG-29b-S, 98.34 ± 8.4; 2A2-SC-S, 98.4 ± 9.2; n = 13) (Figure 2B). Similar to the OSU-CLL cell line, 2A2-miR-29b-ILP significantly downregulated DNMT1, DNMT3A, and SP1 compared with 2A2-scramble-ILP miR control 48 hours posttreatment in primary CLL cells, whereas IgG-miR-29b-ILP exhibited limited effect (Figure 2C; supplemental Figure 4). Moreover, messenger RNA (mRNA) expression analysis of DNMT1, DNMT3A, and SP1 of primary CLL samples from 4 representative patients revealed significant reduction in each of these mRNAs with 2A2-miR-29b-ILP compared with the 2A2-scramble-ILP or IgG-miR-29b-ILP control groups (Figure 2D). Thus, DNMT3A mRNA expression decreased to ∼40% by 2A2-miR-29b-ILP treatment compared with the scramble control, whereas 2A2-ILP delivery enhanced the miR-29b effect (2A2-scramble-ILP, 1.24 ± 0.04; 2A2-miR-29b-ILP, 0.49 ± 0.06; IgG-miR-29b-ILP, 0.83 ± 0.02; P = .0003; n = 4).
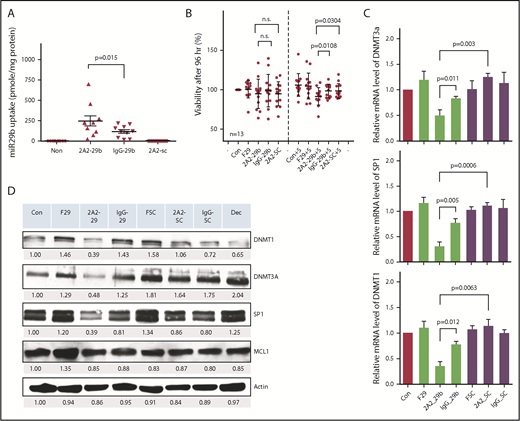
Determination of 2A2-miR-29b-ILP treatment and its downstream targets in primary and cell lines of CLL cells. (A) Treatment of ROR1+ primary CLL with 2A2-miR-29b-ILP for 24 hours showed twofold higher efficiency of ROR1-targeted uptake of miR-29b than nontargeted IgG-miR-29b-ILP control (P = .015; n = 9). (B) The long-term treatment of 2A2-miR-29b-ILP suppressed the cell viability by ∼10% after 96 hours under the coculture of HS-5 stromal cells (n = 13). (C) 2A2-miR-29b-ILP significantly downregulated DNMT1, DNMT3A, and SP1 compared with 2A2-scramble-ILP control 48 hours posttreatment in primary CLL cells, whereas IgG-miR-29b-ILP exhibited limited effect. (D) mRNA expression of DNMT1, DNMT3A, and SP1 of primary CLL samples from 4 patients revealed significant reduction in each of these mRNAs with 2A2-miR-29b-ILP compared with 2A2-scramble-ILP or IgG-miR-29b-ILP control groups. F29, free-form of miR-29b; n.s., not significant; SC, scrambled miR.
Determination of 2A2-miR-29b-ILP treatment and its downstream targets in primary and cell lines of CLL cells. (A) Treatment of ROR1+ primary CLL with 2A2-miR-29b-ILP for 24 hours showed twofold higher efficiency of ROR1-targeted uptake of miR-29b than nontargeted IgG-miR-29b-ILP control (P = .015; n = 9). (B) The long-term treatment of 2A2-miR-29b-ILP suppressed the cell viability by ∼10% after 96 hours under the coculture of HS-5 stromal cells (n = 13). (C) 2A2-miR-29b-ILP significantly downregulated DNMT1, DNMT3A, and SP1 compared with 2A2-scramble-ILP control 48 hours posttreatment in primary CLL cells, whereas IgG-miR-29b-ILP exhibited limited effect. (D) mRNA expression of DNMT1, DNMT3A, and SP1 of primary CLL samples from 4 patients revealed significant reduction in each of these mRNAs with 2A2-miR-29b-ILP compared with 2A2-scramble-ILP or IgG-miR-29b-ILP control groups. F29, free-form of miR-29b; n.s., not significant; SC, scrambled miR.
We also observed the suppression of SP1 mRNA by 2A2-miR-29b-ILP treatment compared with controls (2A2-scramble-ILP, 1.10 ± 0.03; 2A2-miR-29b-ILP, 0.30 ± 0.05; IgG-miR-29b-ILP, 0.77 ± 0.05; P = .0006; n = 4). Moreover, DNMT1, a downstream target of SP1 transcriptional regulation, was also downregulated to ∼30% by 2A2-miR-29b-ILP treatment compared with the 2A2-scramble-ILP control (2A2-scramble-ILP, 1.13 ± 0.07; 2A2-miR-29b-ILP, 0.35 ± 0.05; IgG-miR-29b-ILP, 0.77 ± 0.03; P = .0063; n = 4). Moreover, to evaluate the potential nonspecific effects of 2A2-miR-29b-ILP, we also used normal B cells that do not express ROR1 and ROR1+ primary CLL patient cells. In contrast to primary CLL B cells, normal B cells expressed low levels of DNMT1, DNMT3A, and SP1 (n = 3) (supplemental Figure 5). Importantly, in contrast to free miR29b, 2A2-miR-29b exhibited minimal activation of human B and T cells from 4 healthy donors after 72 hours of treatment (n = 4) (supplemental Figure 6).
ROR1-targeted delivery of miR-29b prolongs survival in the hROR1+ TCL1 mouse model
To evaluate the in vivo effect of ROR1-targeted miR-29b on CLL leukemic cells, we used the Eμ-hROR1 transgenic mouse model expressing human ROR1 (hROR1) exclusively in B cells.15 2A2-ILPs loaded with FAM-ODN fluorescent dye exhibited selective binding of 2A2-ILPs to hROR1-expressing splenocytes ex vivo (Figure 3A). The ROR1-targeted delivery of miR-29b also downregulated DNMT1 and DNMT3A in hROR1-expressing splenocytes by 48 hours, compared with miR-29b–encapsulated control IgG-ILP delivery formulation after ex vivo treatment (Figure 3B). To evaluate the in vivo effect of miR-29b delivery to leukemic B cells, we used the hROR1+ murine leukemic cell engraftment model. Engraftment of leukemic splenocytes from Eμ-ROR1-TCL1 double-transgenic mice into C57BL/6 mice resulted in aggressive leukemia. C57BL/6 recipient mice that exhibited >5% hROR1+B220+CD5+ leukemic B cells in peripheral white blood cells were randomly assigned to 2A2-miR-29b-ILP, IgG-miR-29b-ILP, 2A2-scramble (SC)-ILP, or vehicle treatment cohorts and were treated intraperitoneally Monday, Wednesday, and Friday every week for 6 weeks with the respective formulations (Figure 3C). Assessment of the leukemic population in peripheral blood by using flow cytometry revealed an ∼50% decrease in CD45+B220+CD5+ leukemic cells, but not CD45+B220+CD5– nonleukemic normal B cells, in 2A2-miR-29b-ILP–treated mice compared with the 2A2-scramble-ILP and IgG-miR-29b-ILP groups (Figure 3D). Moreover, the ROR1-targeted delivery of miR-29b prolonged survival of hROR1-expressing leukemia cell–engrafted mice. Thus, in this aggressive mouse model of CLL, the median survival for the engrafted mice treated with phosphate-buffered saline, 2A2-scramble-ILP, IgG-miR-29b-ILP, and 2A2-miR-29b-ILP were 17, 21, 27, and 36 days, respectively. Compared with the 2A2-scramble-ILP treatment (21 days, n = 7), the 2A2-miR-29b-ILP–treated mice lived significantly longer (36 days, n = 9; P = .0019). Moreover, miR-29b delivered by 2A2-ILP (n = 9) prolonged survival of mice significantly over IgG-miR-29b-ILP control (n = 7) (36 vs 27 days; P = .027). Despite these differences in survival, the mice in all 4 groups ultimately died of CLL-like disease, confirming leukemic engraftment (Figure 3E).
![Figure 3. Antileukemic activity of miR-29b with ROR1-targeted delivery of 2A2-ILP in human ROR1 × TCL1 mouse model. (A) 2A2-ILP labeled with FAM-ODN exhibited selective binding of 2A2-ILP to hROR1-expressed splenocytes ex vivo. (B) The 2A2-miR-29b-ILP downregulated DNMT1 and DNMT3A in hROR1-expressed splenocytes by 48 hours, compared with IgG-miR29-bILP control after ex vivo treatment. (C) Engraftment of leukemic splenocytes from Eμ-ROR1-TCL1 double transgenic mice into C57BL/6 mice produced aggressive leukemia. These mice were randomly assigned into 2A2-miR-29b-ILP, IgG-miR-29b-ILP, 2A2-scramble (SC)-ILP, or vehicle treatment cohorts and treated 3 days a week (Monday/Wednesday/Friday every week intraperitoneally [i.p.]). (D) Decreased B220+CD5+ leukemic cells (blue region) by ∼50%, but not nonleukemic B220+CD5– normal B-cell population (purple region), in 2A2-miR-29b-ILP–treated mice compared with the 2A2-scramble-ILP and IgG-miR-29b-ILP groups. (E) 2A2-miR-29b-ILP treatment (36 days, n = 9; P = .0019; log-rank test) prolonged mice survival significantly compared with the 2A2-scramble-ILP treatment (21 days; n = 7) and IgG-miR-29b-ILP control (27 days; n = 7).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/5/10.1182_blood.2018882290/3/m_blood882290f3.png?Expires=1771246206&Signature=4~eIC2dPNct58bnm2xmgFQ8eDUNuVVm1s75AID2C00zhTUxbj0ZYJS~ubAzgbdcdYkaXHl~52~9Z8JOJc~~qst0Mi0Pbt~3crmdxMVpYKrndtn2CGmIt6ZMuFM0R-vG0sKIGEvPu3Br4IS3GkGtN3NWXiMSUFX-~4Tk175--VgulKLXCjTz8gCTFjO1ujZ9Eqc9WvnF6CfOF3vsARYRofCLdR8ZAz~2zLW8IWW4Q0FZGDtkrN56YLBVaZoHMNfypgqRE2HUhlSaE~xHpCaFN4tVbRtEcb9me7oWgqjcOyDBjYEJI5CV~CCwUCizmqyzpy~7fWuz1Qs3-0gtFmmyG7Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Antileukemic activity of miR-29b with ROR1-targeted delivery of 2A2-ILP in human ROR1 × TCL1 mouse model. (A) 2A2-ILP labeled with FAM-ODN exhibited selective binding of 2A2-ILP to hROR1-expressed splenocytes ex vivo. (B) The 2A2-miR-29b-ILP downregulated DNMT1 and DNMT3A in hROR1-expressed splenocytes by 48 hours, compared with IgG-miR29-bILP control after ex vivo treatment. (C) Engraftment of leukemic splenocytes from Eμ-ROR1-TCL1 double transgenic mice into C57BL/6 mice produced aggressive leukemia. These mice were randomly assigned into 2A2-miR-29b-ILP, IgG-miR-29b-ILP, 2A2-scramble (SC)-ILP, or vehicle treatment cohorts and treated 3 days a week (Monday/Wednesday/Friday every week intraperitoneally [i.p.]). (D) Decreased B220+CD5+ leukemic cells (blue region) by ∼50%, but not nonleukemic B220+CD5– normal B-cell population (purple region), in 2A2-miR-29b-ILP–treated mice compared with the 2A2-scramble-ILP and IgG-miR-29b-ILP groups. (E) 2A2-miR-29b-ILP treatment (36 days, n = 9; P = .0019; log-rank test) prolonged mice survival significantly compared with the 2A2-scramble-ILP treatment (21 days; n = 7) and IgG-miR-29b-ILP control (27 days; n = 7).
Antileukemic activity of miR-29b with ROR1-targeted delivery of 2A2-ILP in human ROR1 × TCL1 mouse model. (A) 2A2-ILP labeled with FAM-ODN exhibited selective binding of 2A2-ILP to hROR1-expressed splenocytes ex vivo. (B) The 2A2-miR-29b-ILP downregulated DNMT1 and DNMT3A in hROR1-expressed splenocytes by 48 hours, compared with IgG-miR29-bILP control after ex vivo treatment. (C) Engraftment of leukemic splenocytes from Eμ-ROR1-TCL1 double transgenic mice into C57BL/6 mice produced aggressive leukemia. These mice were randomly assigned into 2A2-miR-29b-ILP, IgG-miR-29b-ILP, 2A2-scramble (SC)-ILP, or vehicle treatment cohorts and treated 3 days a week (Monday/Wednesday/Friday every week intraperitoneally [i.p.]). (D) Decreased B220+CD5+ leukemic cells (blue region) by ∼50%, but not nonleukemic B220+CD5– normal B-cell population (purple region), in 2A2-miR-29b-ILP–treated mice compared with the 2A2-scramble-ILP and IgG-miR-29b-ILP groups. (E) 2A2-miR-29b-ILP treatment (36 days, n = 9; P = .0019; log-rank test) prolonged mice survival significantly compared with the 2A2-scramble-ILP treatment (21 days; n = 7) and IgG-miR-29b-ILP control (27 days; n = 7).
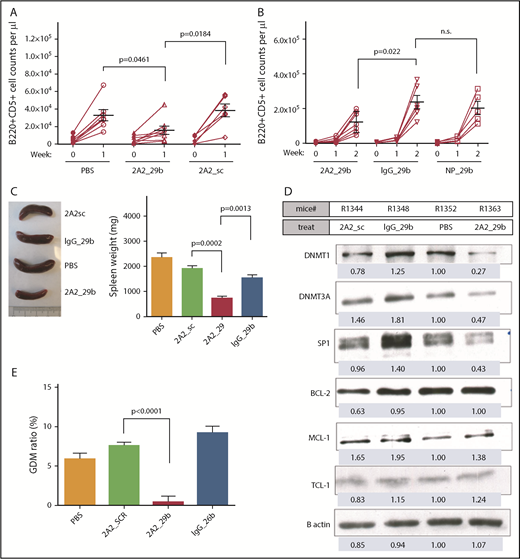
To further evaluate the antitumor efficacy of targeted delivery of miR-29b in vivo, we monitored the total leukemic population in our engrafted mouse model; miR-29b treatment showed significant therapeutic efficacy, as evidenced by a >50% decrease in circulating leukemic B220+CD5+ cells in peripheral blood (2A2-miR-29b-ILP, 15.7 ± 4.6 × 103 cells/μL; n = 8) vs (2A2-scramble-ILP, 38.4 ± 7.3 × 103 cells/μL; n = 6 [P = .0184]) (Figure 4A). Evaluation of leukemic burden in peripheral blood 2 weeks posttreatment showed that ROR1-targeted delivery via 2A2-ILP effectively limited leukemic cell numbers (122.4 ± 23.2 × 103 cells/μL; n = 7) compared with nonselective IgG-miR-29b-ILP (237.1 ± 40.0 × 103 cells/μL; n = 6; P = .022) (Figure 4B). Consistent with the reduction in circulating leukemia cells, reduced splenomegaly was observed in hROR1 × TCL1 cell engrafted mice by 2 weeks posttreatment with 2A2-miR-29b-ILP compared with those that received 2A2-scramble-ILP control (2A2-miR-29b-ILP, 734.3 ± 90.2; 2A2-scramble-ILP, 1918 ± 110 mg; P = .0002; n = 4) (Figure 4C). Immunoblotting of splenocytes from treated mice revealed downregulation of miR-29b targets such as DNMT1, DNMT3A, and SP1 with the 2-week treatment. Due to the absence of 3′ untranslated region of TCL1 mRNA in our Eu-Tcl1 transgenic model, we observed no downregulation of TCL1, a known miR-29b target (Figure 4D; supplemental Figure 7). Moreover, a 2-week treatment of mice with miR-29b-2A2-ILP significantly reduced GDM by 20-fold compared with scramble miR-2A2-ILP control in splenocytes (2A2-miR-29b-ILP, 0.4 ± 0.76%; 2A2-scramble-ILP, 7.54 ± 0.46%; P < .0001; n = 4) (Figure 4E). Furthermore, to determine if the 2A2-ILP formulation will lead to nonspecific immune activation in vivo, we treated immunocompetent human ROR1 × TCL1 transgenic mice with vehicle, 2A2-miR-29b-ILP, or free-form miR-29b. No detectable in vivo activation of mouse B and T cells was observed with 2A2-miR-29b-ILP treatment (supplemental Figure 8).
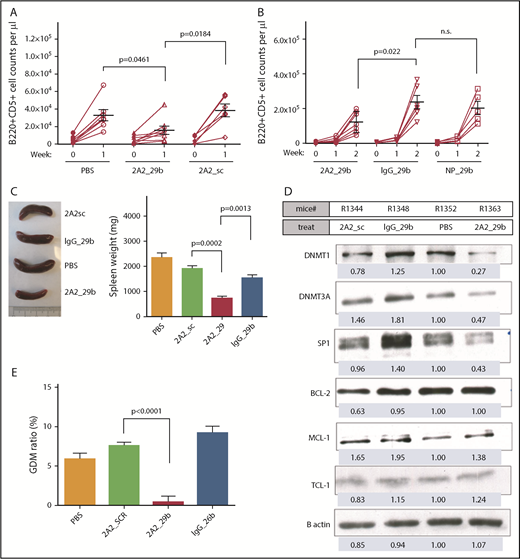
Antileukemic activity of miR-29b reduced the leukemic progression in the hROR1 × TCL1 mouse model. (A) The miR-29b treatment (2A2-miR-29b-ILP, 15.7 ± 4.6 × 1000 cells/μL; n = 8) showed significant therapeutic efficacy as evidenced by decreased circulating leukemic B220+CD5+ cells by >50% in peripheral blood (2A2-scramble-ILP, 38.4 ± 7.3; n = 6; P = .0184). (B) Evaluation of leukemic burden in peripheral 2-week posttreatment revealed ROR1-targeted delivery via 2A2-ILP effectively limited leukemic cell numbers (122.4 ± 23.2 × 1000 cells/μL; n = 7) compared with nonselective IgG-miR-29b-ILP (237.1 ± 40.0 × 1000 cells/μL; n = 6; P = .022). (C) Reduced splenomegaly in hROR1 × TCL1 mice by 2 weeks posttreatment with 2A2-miR-29b-ILP compared with the 2A2-scramble-ILP control (P = .0002; n = 4). (D) Immunoblot analysis of splenocytes from treated mice revealed downregulation of miR-29b targets such as DNMT1, DNMT3A, and SP1 at protein level after 2 weeks of in vivo treatment. (E) Significant reduction in GDM in mouse splenocytes 2 weeks posttreatment with 2A2-miR-29b-ILP compared with 2A2-scramble-ILP control (P < .0001; n = 4).
Antileukemic activity of miR-29b reduced the leukemic progression in the hROR1 × TCL1 mouse model. (A) The miR-29b treatment (2A2-miR-29b-ILP, 15.7 ± 4.6 × 1000 cells/μL; n = 8) showed significant therapeutic efficacy as evidenced by decreased circulating leukemic B220+CD5+ cells by >50% in peripheral blood (2A2-scramble-ILP, 38.4 ± 7.3; n = 6; P = .0184). (B) Evaluation of leukemic burden in peripheral 2-week posttreatment revealed ROR1-targeted delivery via 2A2-ILP effectively limited leukemic cell numbers (122.4 ± 23.2 × 1000 cells/μL; n = 7) compared with nonselective IgG-miR-29b-ILP (237.1 ± 40.0 × 1000 cells/μL; n = 6; P = .022). (C) Reduced splenomegaly in hROR1 × TCL1 mice by 2 weeks posttreatment with 2A2-miR-29b-ILP compared with the 2A2-scramble-ILP control (P = .0002; n = 4). (D) Immunoblot analysis of splenocytes from treated mice revealed downregulation of miR-29b targets such as DNMT1, DNMT3A, and SP1 at protein level after 2 weeks of in vivo treatment. (E) Significant reduction in GDM in mouse splenocytes 2 weeks posttreatment with 2A2-miR-29b-ILP compared with 2A2-scramble-ILP control (P < .0001; n = 4).
MiR-29b treatment results in cell cycle arrest that is dependent on decreased SP1 and increased p21 induction
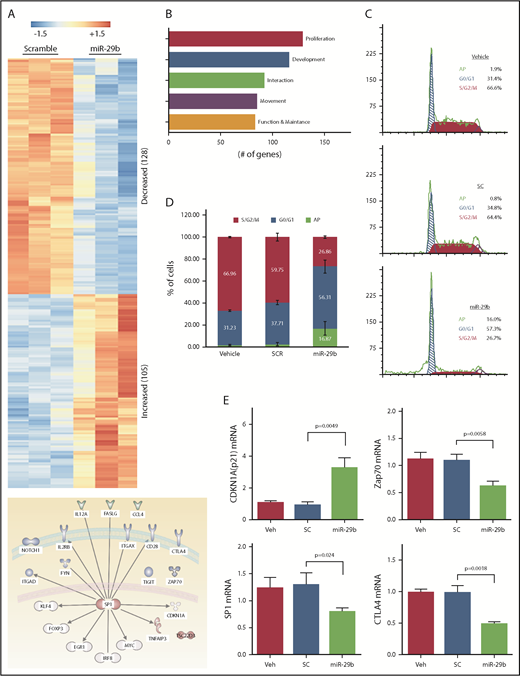
To investigate the mechanistic basis of miR-29b–mediated reductions in leukemic burden and disease progression, we performed RNA-seq analysis of purified splenic CD19+ B lymphocytes from the mice treated with 2A2-miR-29b-ILP or 2A2-control scramble-ILP. Transcript analysis revealed at least 233 differentially expressed genes between 2A2-miR-29b-ILP vs 2A2-scramble-ILP control (Figure 5A; supplemental Table 2). Expression of 128 genes was decreased, whereas 105 genes were upregulated. These genes represent modulators of cell growth, proliferation, development, cell–cell interaction, and cell migration. Interestingly, the majority of the differentially expressed genes are related to cell proliferation (Figure 5B). Consistent with a role for miR29b in deregulation of cell proliferation, cell cycle analysis revealed accumulation of leukemic cells in G0/G1 with a concomitant decrease of cells in G2/S/M in vivo in the 2A2-miR-29b ILP–treated group compared with the control 2A2-scramble-ILP–treated group (2A2-scramble-ILP, 37.7 ± 2.0% [G0/G1], 59.8 ± 3.5% [G2/S/M] vs 2A2-miR-29b-ILP, 56.3 ± 5.9% [G0/G1], 26.9 ± 1.1% [G2/S/M]; G0/G1, P = .016; G2/S/M, P = .0002; n = 4) (Figure 5C). Moreover, apoptotic cells were also increased in the miR-29b treatment group (2A2-scramble-ILP, 2.55 ± 0.8%; 2A2-miR-29b-ILP, 16.9 ± 3.1%) (Figure 5D). Independent validation of RNA-seq results by using quantitative real-time polymerase chain reaction confirmed a decrease in SP1, ZAP70, and CTLA-4 and an increase in cell cycle inhibitor p21 mRNA transcription upon 2A2-miR-29b-ILP treatment compared with vehicle and 2A2-scramble-ILP controls (Figure 5E).
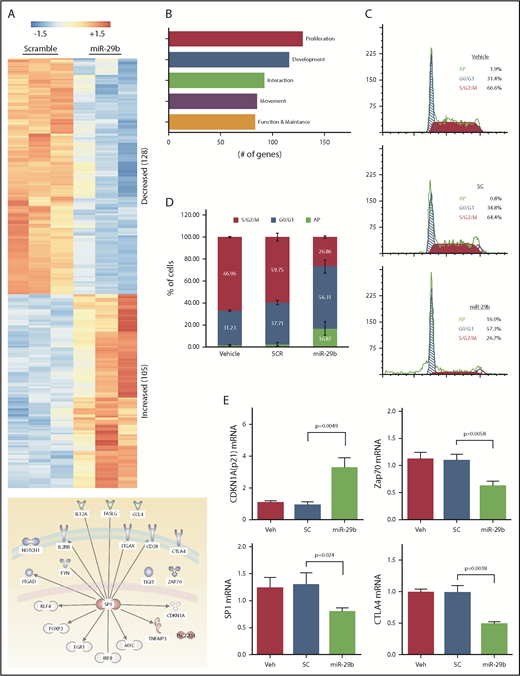
Long-term treatment with miR-29b suppressed proliferation of leukemic cells in vivo. (A) RNA-seq from purified splenic CD19+ B lymphocytes from the mice treated with 2A2-miR-29b-ILP or 2A2-control scramble-ILP. Differential expression of genes revealed at least 233 differentially expressed genes between miR-29b vs scramble control (128 genes decreasing and 105 genes increasing). (B) A total of 128 of the 233 differentially expressed genes represent cellular growth and proliferation. (C-D) Cell cycle analysis of CD19+ B cells from the in vivo 2A2-miR-29b ILP–treated group compared with the control 2A2-scramble-ILP–treated group. Panel C shows representative data of vehicle, 2A2-scramble-ILP2 (SC), and 2A2-miR29b ILP (mR-29b) treated groups, and panel D presents the summarized data (P = .0002; n = 4). Panel D also shows increased apoptotic cells in the 2A2-miR29b-ILP treatment group (scramble, 2.55 ± 0.8%; miR-29b, 16.9 ± 3.1%). (E) Validation of decreased expression of SP1, ZAP70, and CTLA-4 mRNA and increased expression of CDKN1a/p21 mRNA in the post–2A2-miR-29b-ILP treatment group confirmed by differential expression of genes.
Long-term treatment with miR-29b suppressed proliferation of leukemic cells in vivo. (A) RNA-seq from purified splenic CD19+ B lymphocytes from the mice treated with 2A2-miR-29b-ILP or 2A2-control scramble-ILP. Differential expression of genes revealed at least 233 differentially expressed genes between miR-29b vs scramble control (128 genes decreasing and 105 genes increasing). (B) A total of 128 of the 233 differentially expressed genes represent cellular growth and proliferation. (C-D) Cell cycle analysis of CD19+ B cells from the in vivo 2A2-miR-29b ILP–treated group compared with the control 2A2-scramble-ILP–treated group. Panel C shows representative data of vehicle, 2A2-scramble-ILP2 (SC), and 2A2-miR29b ILP (mR-29b) treated groups, and panel D presents the summarized data (P = .0002; n = 4). Panel D also shows increased apoptotic cells in the 2A2-miR29b-ILP treatment group (scramble, 2.55 ± 0.8%; miR-29b, 16.9 ± 3.1%). (E) Validation of decreased expression of SP1, ZAP70, and CTLA-4 mRNA and increased expression of CDKN1a/p21 mRNA in the post–2A2-miR-29b-ILP treatment group confirmed by differential expression of genes.
SP1 is overexpressed in multiple cancer types associated with poor prognosis.17,18 CLL has been shown to overexpress Sp1,19 and knockdown of Sp1 results in suppression of G1/S phase transition by inducing p21 in carcinoma and gastric cancer.20,21 To mechanistically determine if miR-29b–mediated deregulation of cell cycle progression is dependent on the Sp1/p21 axis, we knocked down Sp1 expression by small interfering RNA (siRNA) in the OSU-CLL cell line and evaluated the effect of 2A2-miR-29b-ILP. Consistent with our observation in the hROR1 × TCL1 mouse model of CLL, 2A2-miR-29b-ILP treatment downregulated Sp1 levels, with concomitant increases in p21 protein expression in the human OSU-CLL cell line, and siRNA-mediated knockdown of SP1 resulted in increased p21 expression in 2A2-scrambled-ILP that was further enhanced with 2A2-mir29b-ILP (Figure 6A).
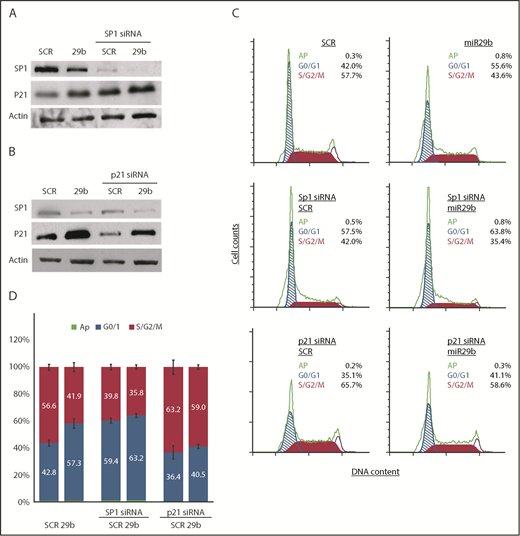
miR-29b suppressed leukemic cell proliferation in SP1/p21–dependent manner. (A) 2A2-miR-29b-ILP (29b) downregulated SP1 and increased p21 expression in the human OSU-CLL cell line (lanes 1 and 2). The siRNA-mediated knockdown of SP1 resulted in increased p21 expression (lanes 3 and 4). (B) Transfection of p21-directed siRNA decreased control (SCR) as well as 2A2-miR29b-ILP (29b) induced p21 protein. (C-D) Accumulation of cells in G0/G1 with concomitant decrease in S/G2/M by miR-29b that is reversed upon SP1 and p21 knockdown in OSU-CLL cells. Panel C shows results from a representative experiment. Panel D presents compiled data from 4 independent experiments.
miR-29b suppressed leukemic cell proliferation in SP1/p21–dependent manner. (A) 2A2-miR-29b-ILP (29b) downregulated SP1 and increased p21 expression in the human OSU-CLL cell line (lanes 1 and 2). The siRNA-mediated knockdown of SP1 resulted in increased p21 expression (lanes 3 and 4). (B) Transfection of p21-directed siRNA decreased control (SCR) as well as 2A2-miR29b-ILP (29b) induced p21 protein. (C-D) Accumulation of cells in G0/G1 with concomitant decrease in S/G2/M by miR-29b that is reversed upon SP1 and p21 knockdown in OSU-CLL cells. Panel C shows results from a representative experiment. Panel D presents compiled data from 4 independent experiments.
Transfection of p21-directed siRNA decreased endogenous p21 protein as well as the 2A2-miR29b-ILP–induced p21 protein (Figure 6B). Importantly, cell cycle analysis of the OSU-CLL cells also revealed accumulation of cells in the G0/G1 phase upon miR-29b treatment. To further assess the miR-29b effect in the absence of Sp1 or p21, we pretreated OSU-CLL cells with specific siRNA of either SP1 or p21 for 24 hours, and then incubated the cells with 2A2-miR-29b-ILP or 2A2-scramble-ILP controls for 48 hours. Cell cycle analysis revealed that miR-29b significantly arrested cells in the G0/G1 phase, which was negated upon Sp1-knockdown (14.9% vs 3.96%; P < .0001) (Figure 6C-D), indicating that miR-29b–induced cell cycle arrest in CLL cells is dependent on SP1. Moreover, we also evaluated the effect of miR-29b on G0/G1 arrest in cells with p21 knockdown. Although miR-29b caused significant arrest of cells in the G0/G1 phase in the control group, p21-siRNA–mediated knockdown rescued the G0/G1 arrest in the p21 knockdown group (4.12% vs 14.9% control; P < .0001), indicating that the cell cycle arrest induced by miR-29b is p21 dependent.
Discussion
Dysregulated miR expression has been implicated in diverse cancers, supporting their role in aberrantly modulating functional targets such as tumor suppressors or oncogenes.22 Despite the potential promise, miR therapeutics have not progressed into the clinical setting, partly due to limitations associated with their short half-life in circulation and the notorious regulatory effects on diverse targets that are not only functionally relevant in the disease establishment and progression but also critical in normal physiology of healthy tissues and organ systems. As a proof of principle, we describe efforts to overcome these limitations using a novel mouse model and a targeted delivery formulation of miR-29b that is directed to ROR1, an oncofetal antigen expressed on CLL cells but not normal B cells.8,15,23 In patients with aggressive CLL, miR-29b is downregulated and contributes to disease pathogenesis.24 In multiple cancers, downregulation of miR-29b is associated with posttranslational and epigenetic regulation of multiple survival genes resulting in enhanced survival of cancer cells, drug resistance, and poor prognosis.25,26 Thus, a therapeutic increase of miR-29b in cancer cells could provide substantial clinical benefit.
The current clinical experience of miR-based cancer therapeutics is limited to the miR-34 family, which is downregulated in many types of cancer.27,28 The miR-34 family received much attention with its regulatory role in p53-mediated apoptosis upon DNA damage by directly targeting several antiapoptotic proteins.29 In 2013, MRX34, a nanoparticle-encapsulated miR-34 mimic (Mirna Therapeutics), first entered a multicenter phase 1 trial in patients with primary liver cancer, small cell lung cancer, lymphoma, melanoma, multiple myeloma, or renal cell carcinoma.2,30,31 At the end of the trial, a therapeutic benefit was observed in certain patients with significant reduction of the miR-34 target mRNAs in their blood samples. However, because of immune-related serious adverse events, the trial was terminated (see www.clinicaltrials.gov, #NCT01829971 for MRX34). Despite attempts to encapsulate the miRs in a lipid carrier to enhance sustainable delivery and accumulation in the tumor microenvironment, the severe immune reactions were discouraging.32 These experiences reiterated the need for targeted delivery of miR formulations with emphasis on selectivity and specificity to avoid possible immune-related toxicities.
Freely circulating synthetic miR drugs are limited by their unpredictable degradation in biofluids and low cellular uptake, whereas encapsulation of miRs within nanocarriers can prolong their half-life and increase their uptake. Although the reticuloendothelial system effect helps therapeutic nanoparticles compared with free drugs to accumulate in tumor sites, the poor selectivity of delivery still results in side effects.33 Therefore, developing a selective delivery system to further enhance cancer cell uptake and limit uptake in normal cells is critical.
Immunoliposomal targeting for B-cell malignancy has focused on CD20, CD19, and CD52, which are widely expressed in multiple cell types of B-cell lineage and other immune system components.34,35 ROR1, a receptor tyrosine kinase-like orphan receptor, is identified as being expressed in >95% of CLL cells but not in normal B cells, thus providing opportunity for a leukemia-specific target for delivery.36 Moreover, higher ROR1 expression in CLL is correlated with high AKT activation, likely contributing to aggressive disease compared with the ROR1-negative cases.23 ROR1-targeted delivery of miR-29b described here is an attractive strategy to specifically target leukemic cells while sparing normal B cells.
Here, we show an efficient and selective delivery via targeting ROR1 to directly increase mature miR-29b levels in target CLL cells in vitro and in vivo. Sustained overexpression of miR-29 in murine B cells by the transgenic Eμ promoter leads to a CD5+CD19+ B-cell malignancy, which precludes the use of free miR-29b for therapeutic purposes and highlights the need to prevent exposure of developing B cells.6 The delivery of miR-29b via our ROR1-targeted 2A2-ILP to leukemic B cells is a significant advancement that will avoid the aforementioned limitations and spare normal B cells. We and others have widely used the Eμ-TCL1 CLL mouse model for in vivo evaluation of treatments for CLL.37 Crossing Eμ-Tcl1 mice to relevant Eμ-driven human CD37 or human ROR1 transgenic lines has faithfully reproduced the aggressive murine CLL but with human markers for therapy evaluation and drug targeting.15,38
The low proliferation rate (<10%) of human circulating B-CLL cells suggests that the splenic microenvironment is required for proliferating, also increasing the difficulty of monitoring in patients. Using Eµ-TCL1 or NZB IRF4+/− transgenic mouse, the leukemic cells exhibit a proliferative preference within the spleen (15% to 20%) than peripheral blood (∼5%). Mimicking the stimulation via ROR1 signaling in aggressive CLL, we crossed ROR1 with TCL1-transgenic mice to further enhance the leukemia growth and resistance to apoptosis, producing a more aggressive disease model upon adoptive transfer via serial transplantation than conventional TCL1-transgenic leukemia. TCL1 is known to be directly regulated by the miR-29 family. Overexpression of TCL1 acts as a coactivator of AKT serine–threonine kinase that is necessary to trigger leukemogenesis. Although miR-29b could directly suppress TCL1 and thus delay disease progression in the transgenic model,39 this possibility is ruled out as the TCL1 transgenic construct used here does not include the miR-29b target 3′ untranslated region. Instead, our in vivo results ascribe the therapeutic benefit of miR-29b to deregulation of the cell cycle rather than inhibition of TCL1 translation. Most importantly, the selective delivery of miR-29b via ROR1 targeting directly increased mature miR-29b levels in the leukemic cells and suppressed DNMT1, DNMT3A, and SP1 levels, leading to epigenetic alterations in vitro and in vivo. Interestingly, miR-29b believed to suppress MCL1 expression in cholangiocarcinoma40 and pancreatic cancer41 has little effect on reducing MCL1 expression in B-cell lineages, either in primary CLL or the TCL1 mouse model. This result is consistent with the mouse model established by Santanam et al,6 as the Eμ-overexpressed miR-29 bypassed the suppressive function to MCL1 expression.
Global, CpG-regional, or gene-specific hypermethylation affecting putative tumor suppressor genes has been reported in CLL.40,42 DNMTs play critical roles in physiological DNA methylation, but their involvement in CLL remains to be clarified.43 Mutations in the coding sequence of DNMT3A are frequently observed in human myeloid and certain lymphoid malignancies,44 and reduction or inactivation of DNMT3A induces a CD5+B220+ CLL-like leukemia in mice.45,46 Interestingly, heterogeneous methylation forms the basis of intratumor methylome variation in CLL.47 In a phase 1 trial of decitabine, patients with CLL exhibited no evidence of DNA hypomethylation at a low dose.48 Here, we report the reduction of GDM via downregulation of DNMTs with long-term miR-29b treatment. Thus, the ROR1-targeted nanoparticle described here may reduce hypermethylation that is selective to CLL, while sparing normal tissues and reducing side effects.
Our findings of DNMTs and Sp1 downregulation in primary CLL cells treated with miR-29b in vitro and in TCL1 leukemic cells in vivo are consistent with the miR-29b/SP1 feedback loop regulating the cell cycle and survival reported in acute myeloid leukemia and multiple myeloma. In multiple myeloma, the phosphatidylinositol 3-kinase/AKT pathway interacts with the miR-29b-Sp1 loop to regulate apoptosis, and high levels of miR-29b in vivo significantly reduced tumorigenic potential.49 In a KIT-driven acute myeloid leukemia model, the Sp1/NF-kB/HDAC/miR-29b network contributes to leukemia progression and can be successfully alleviated by synthetic miR-29b treatment.50 Moreover, SP1 is upregulated in the CLL-associated B-cell receptor signaling pathway,19 suggesting that overexpression of SP1 induces expression and activity of protein kinase CβII, which is critical for CLL cell growth.
Our in vivo findings showing an miR-29b–mediated decrease in SP1 and an increase in p21 expression in ROR1+ TCL1-overexpressed CLL cells is consistent with a suppressive role of SP1 in p21 regulation. This finding is further confirmed by reversal of miR-29b–mediated SP1 and p21-dependent cell cycle arrest upon SP1 knockdown. Moreover, knockdown of p21 significantly decreased the effect of miR-29 on cell cycle arrest, supporting the role of p21 in miR-29b-SP1–mediated cell cycle regulation.
In conclusion, we show the antitumor effect of selectively delivering miR-29b to CLL cells. We found that ROR1-targeted delivery can efficiently increase low levels of miR-29b both in vitro and in vivo and achieve cell cycle arrest of leukemic cells via a mechanism involving Sp1 and p21. Based on our results, an miR-29b immuno-nanoparticle may warrant further evaluation for potential clinical applications in CLL and other cancers resistant to DNA hypomethylating agents.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to the patients with CLL who generously supported this research by donating their blood. They also thank David Lucas for critical review of the manuscript. The authors gratefully acknowledge The Ohio State University Comprehensive Cancer Center Leukemia Tissue Bank Shared Resource for providing patient samples and the Genetically Engineered Mouse Modeling Shared Resource for generation of the transgenic mice. These facilities are supported by the National Institutes of Health, National Cancer Institute (P30 CA016058).
This work was supported by the National Institutes of Health, National Cancer Institute (R01 CA135332, CA159296, CA165469, CA197844, P01 CA95426, and R35 CA197734), the National Science Foundation (EEC-0914790), the D. Warren Brown Foundation, the Four Winds Foundation, the Sullivan Chronic Lymphocytic Leukemia Research Fund, and the Connie Brown CLL Foundation.
Authorship
Contribution: C.-L.C. planned, designed, and performed the majority of the in vitro and in vivo research, analyzed data, and drafted the initial manuscript; S.G. and X.H. synthesized and characterized ROR1-targeted nanoparticles; F.W.F. and R.M. generated and characterized the ROR1 and ROR1 × TCL1 double transgenic mice; Z.X. and M.A.P. assisted with quantitation of GDM and miR-29b; P.S.Y. performed RNA-seq data acquisition, and P.S.Y., L.A.W., and R.B. conducted the analysis; X.M.M. participated in experimental design and statistical analysis; J.C.B. contributed to the CLL patient care, patient sample acquisition, characterization, and experimental design; S.B., C.R., and G.M. provided necessary reagents of miRs and antibodies; N.M. and L.J.L. oversaw the study, sought funding, and participated in experimental design and data interpretation; and all authors contributed to review, editing and approval of the final manuscript version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Natarajan Muthusamy, Division of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, 455E OSUCCC, Bldg. 410 West 12th Ave, Columbus, OH 43210; e-mail: raj.muthusamy@osumc.edu.


![Figure 3. Antileukemic activity of miR-29b with ROR1-targeted delivery of 2A2-ILP in human ROR1 × TCL1 mouse model. (A) 2A2-ILP labeled with FAM-ODN exhibited selective binding of 2A2-ILP to hROR1-expressed splenocytes ex vivo. (B) The 2A2-miR-29b-ILP downregulated DNMT1 and DNMT3A in hROR1-expressed splenocytes by 48 hours, compared with IgG-miR29-bILP control after ex vivo treatment. (C) Engraftment of leukemic splenocytes from Eμ-ROR1-TCL1 double transgenic mice into C57BL/6 mice produced aggressive leukemia. These mice were randomly assigned into 2A2-miR-29b-ILP, IgG-miR-29b-ILP, 2A2-scramble (SC)-ILP, or vehicle treatment cohorts and treated 3 days a week (Monday/Wednesday/Friday every week intraperitoneally [i.p.]). (D) Decreased B220+CD5+ leukemic cells (blue region) by ∼50%, but not nonleukemic B220+CD5– normal B-cell population (purple region), in 2A2-miR-29b-ILP–treated mice compared with the 2A2-scramble-ILP and IgG-miR-29b-ILP groups. (E) 2A2-miR-29b-ILP treatment (36 days, n = 9; P = .0019; log-rank test) prolonged mice survival significantly compared with the 2A2-scramble-ILP treatment (21 days; n = 7) and IgG-miR-29b-ILP control (27 days; n = 7).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/5/10.1182_blood.2018882290/3/m_blood882290f3.png?Expires=1771315915&Signature=KlWLswAuDHWPHrBMJbH5uX3p7jv4deaZ5j1J9Kemjcej9~PkkClcf~hadQ4fUpkdTLcTfPWzbbJdsUmqeNI85A4HhuJEZET3tijxToofPPVEoaSyP8twSDE5QspjQIlIZUBUyLWrWLNxP7k1eMY20y3H-ORqnBSqYbgiX7fKmct-mOGtxBhhHltVp9RYd21m8-TZQoTJKD6QB43Qo6PZDCJ5YEU0~kcFN10ttPKeBwnOgnJtRCvZe8cA6qGDObBxn~ZUa4~vuUcbFlAigoMj-D44974Tfg4JrbpMBfmTwQMnwuf51tp43xGfIACZ7L0HjLgH-3fqSaPpnhgrN3qPIw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)