Key Points
CALR mutations are prevalent in the general population but are much less frequent compared with the estimated JAK2 V617F prevalence.
JAK2 V617F and CALR mutations in the general population are linked to a distinct blood count profile, also in the absence of MPN diagnosis.

Abstract
The JAK2 V617F and calreticulin mutations (CALR) are frequent within myeloproliferative neoplasms (MPNs). JAK2 V617F has been detected in the general population, but no studies have previously investigated the CALR prevalence. Thus, we aimed to determine the CALR and JAK2 V617F population prevalence and assess the biochemical profile and lifestyle factors in mutation-positive individuals with and without MPN. 19 958 eligible participants, enrolled from 2010-2013, from the Danish General Suburban Population Study were screened for JAK2 V617F and CALR by droplet digital polymerase chain reaction with (3.2%) mutation positives of which 16 (2.5%) had MPN at baseline. Of 645 participants, 613 were JAK2 V617F positive, and 32 were CALR positive, corresponding to a population prevalence of 3.1% (confidence interval [CI], 2.8-3.3) and 0.16% (CI, 0.11-0.23), respectively. Increasing age, smoking, and alcohol were risk factors for the mutations. JAK2 V617F positives with and without MPN presented elevated odds for prevalent venous thromboembolism. The odds ratio for a diagnosis of MPN per percentage allele burden was 1.14 (95% CI, 1.09-1.18; P = 1.6 × 10−10). Mutation positives displayed higher blood cell counts than nonmutated participants, and 42% of mutation positives without MPN presented elevation of ≥1 blood cell counts; 80 (13%) even presented blood cell counts in accordance with current MPN diagnostic criteria. In conclusion, we present a novel population prevalence of CALR and a JAK2 V617F prevalence that is 3 to 30 times higher compared with less sensitive methods. Mutation-positive non-MPNs with elevated blood cell counts raise concerns of MPN underdiagnosis in the population.
Introduction
The driver mutation Janus kinase 2 V617F (JAK2 V617F) and calreticulin mutations (CALR) are frequent within the chronic Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs),1,2 and they constitute one of the major diagnostic criteria of MPNs.3
JAK2 V617F has also been detected in healthy volunteers4,5 and patients with cardiovascular diseases,6 venous thromboses,7 splanchnic vein thromboses,8 and apoplexias,9 all vascular complications associated with MPN. Few studies have assessed CALR in other disease entities,9-15 with only rare detection of CALR in patients with splanchnic vein thrombosis14 and cerebral venous thrombosis.15 JAK2 V617F is also more prevalent among smokers,16 and smoking has been suggested to be a risk factor for MPN development17-21 and other related myeloid neoplasias22-24 due to the chronic inflammatory stimulus.
Few studies have investigated the prevalence of JAK2 V617F in the general population25 or larger cohorts.26-29 A Danish general population study found the JAK2 V617F prevalence to be 0.1% based on final quantification by real-time quantitative polymerase chain reaction (PCR).25 By whole-exome sequencing, 2 research groups have assessed clonal hematopoiesis of indeterminate potential (CHIP) in larger cohorts unselected for hematologic phenotype at the time of blood sampling and found JAK2 V617F to be prevalent in 0.18%26 and 0.19%,27 respectively. Also in cohorts unselected for blood disorders, McKerrell et al detected a JAK2 V617F prevalence of 0.61% by next-generation sequencing.28 Among random hospital blood samples, Xu et al detected a prevalence of 0.94% found by a nested allele-specific PCR method.29 To our knowledge, no previous studies have estimated the population prevalence of CALR.
Thus, the aims of this cross-sectional study are to determine the prevalence of CALR type 1 and 2 as well as JAK2 V617F in the Danish General Suburban Population Study (GESUS) by use of a highly sensitive method and to characterize these individuals’ biochemical profile and lifestyle in relation to their mutational status.
Methods
Participants
The study population consisted of GESUS (N = 21 205), where participants had DNA stored in a biobank for future research after proper consent. GESUS invited all Danish citizens irrespective of health status in the municipality of Naestved aged >30 years and 25% of citizens aged 20 to 30 years from January 2010 to October 2013.30 The population study consisted of a health examination, biochemical measurements, and a questionnaire about health and lifestyle (for details, see supplemental Methods, available on the Blood Web site).
Study protocols were approved by the Regional Committee on Health Research Ethics (SJ-452) and the Danish Data Protection Agency (REG-50-2015) and comply with the Declaration of Helsinki.
Molecular screening design
The screening was performed by a pooled multiplex droplet digital PCR (ddPCR) assay (supplemental Figure 1) with DNA from GESUS. DNA from 4 participants was pooled and analyzed by a JAK2 V617F and a CALR type 1 and type 2 assay. Both assays were multiplexed with wild-type. If mutation positive, the 4 samples were reanalyzed separately for either JAK2 V617F or CALR to identify the positive sample(s) and quantify the mutant allele burden.
See supplemental Methods for details about applied ddPCR conditions, DNA, primers, and probes.
Level of detection
The sensitivity of the assays was calculated to be 0.009% for JAK2 V617F and 0.01% for CALR type 1 and 2. For details, see supplemental Methods.
Elevation of blood cells
Elevation of peripheral blood cells was defined according to the following current regional ranges: hemoglobin >15.3 g/dL (female) or >16.9 g/dL (male); hematocrit >0.46 (female) or >0.50 (male); erythrocytes >5.2 × 1012/L (female) or >5.7 × 1012/L (male); leukocytes >8.8 × 109/L; neutrophils >7.0 × 109/L; monocytes >0.7 × 109/L; eosinophils ≥0.5 × 109/L; basophils ≥0.1 × 109/L; lymphocytes >3.5 × 109/L; and thrombocytes >390 × 109/L.
MPNs
From the electronic medical records, we manually retrieved a diagnosis of MPN present at time of participation in GESUS on all mutation-positive participants. Thus, the diagnosis was made prior to GESUS in standard clinical settings by clinical hematologists according to current international guidelines at the time of diagnosis. MPN subdiagnoses included in this study were polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), prefibrotic myelofibrosis, and unclassifiable MPN.
Study design and statistics
In this cross-sectional study, we detected the prevalence of the mutations in the general population, and basic characteristics and blood tests were compared between mutation-positive participants and the nonmutated background population.
Second, we assessed the JAK2 V617F positives stratified according to allele burden <1% and ≥1%. We also analyzed mutation positives stratified in MPNs and non-MPNs vs nonmutated to assess whether any differences between mutation positives and nonmutated participants were only due to the prevalent MPN disease among the mutation positives.
Thirdly, multivariable logistic regression was performed to determine the odds ratio for thrombotic/ischemic events (self-reported) prior to GESUS for JAK2 V617F–positive non-MPNs and MPNs compared with nonmutated. To assess whether associations were mediated by blood cell counts, both a model with and a model without blood cell counts were included.
Finally, we assessed the possible associations of lifestyle variables (smoking, alcohol, and body mass index) and mutational status with the latter as the response variable. To bypass any reverse causation, we repeated these analyses in a subpopulation with no self-reported history of smoking-related diseases (no acute myocardial infarction, ischemic cerebrovascular disease, ischemic heart disease, cancer, and diabetes and no use of asthma/bronchitis medication, antihypertensive medication, and diabetes medication).
Stata/SE 14 was used for statistical analyses. P < .05 was considered statistically significant, and confidence intervals (CIs) were 95%. Linear regression was used in analyses of continuous dependent variables with logistic transformation applied when relevant. Statistically significant differences among groups were not considered clinically relevant if presented mean values were identical. Logistic regression was used in analyses with a binary dependent variable. All regression analyses were adjusted for age and gender except for estimated glomerular filtration rate (eGFR). Pearson χ2 test was used in comparisons of 2 multicategorical variables. If double mutated (concurrent JAK2 V617F and CALR positive), the individual would be included in comparative statistics among the JAK2 V617F or CALR positives according to the highest allele burden present.
Results
Prevalence of JAK2 V617F and CALR
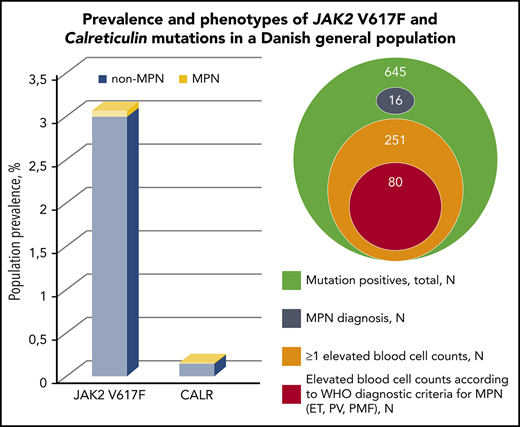
Screening for JAK2 V617F and CALR was performed in 19 958 GESUS participants. Six hundred forty-five participants (3.2%) were mutation positive, and 16 of these (2.5%) had MPN at enrolment. JAK2 V617F was found in 613 participants, corresponding to a population prevalence of 3.1% (CI, 2.8-3.3). CALR was found in 32 participants, corresponding to a population prevalence of 0.16% (CI, 0.11-0.23), with the type 1 mutation constituting 75% (supplemental Figure 2). The ratio of JAK2 V617F to CALR was 19:1. The majority of mutation-positive participants had allele burdens <1% (Table 1). Three participants were homozygous in JAK2 V617F (allele burdens all >90% and all had a MPN diagnosis), whereas none of the CALR positives had allele burden >50%. Two participants were double mutated (both JAK2 V617F and CALR type 1). If applying the detection limits from previous studies on JAK2 V617F prevalence in larger cohorts outside MPN25-29 to our JAK2 V617F data, we obtain a similar prevalence (supplemental Table 1). According to smoking status, the mutations were most prevalent among current smokers (3.5%), followed by former smokers (3.3%), and lowest among never smokers (3.1%).
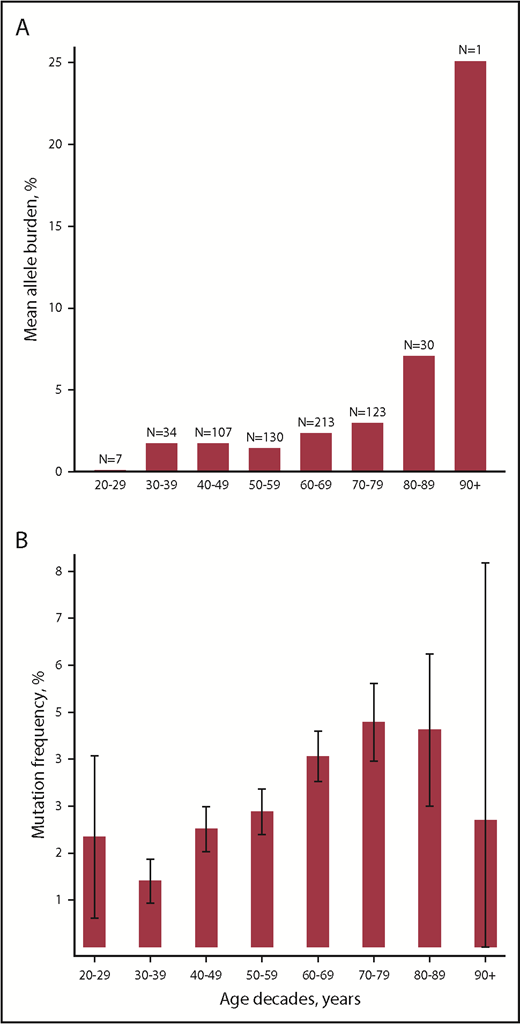
Age was positively associated with allele burden (β coefficient, 1.01; 95% CI, 1.00-1.03; P = .02) and with presence of mutation with odds ratio of 1.02 (95% CI, 1.02-1.03; P = 7.1 × 10−15) as also depicted in Figure 1.
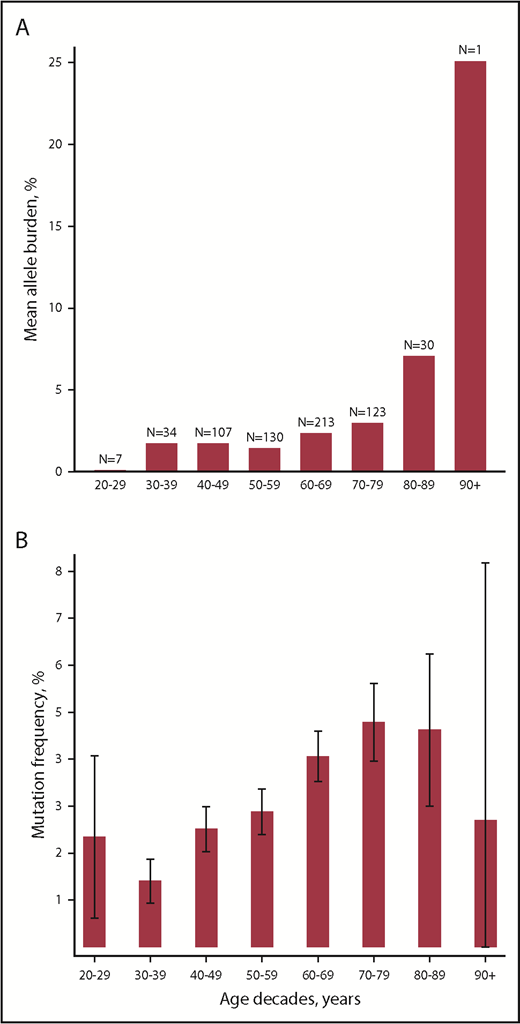
Associations between age and MPN driver mutations. (A) Mean allele burden per age decade. (B) Mutation frequency per age decade. Caped spikes represent 95% CIs.
Associations between age and MPN driver mutations. (A) Mean allele burden per age decade. (B) Mutation frequency per age decade. Caped spikes represent 95% CIs.
The distributions of mutation types and allele burdens according to MPN subdiagnoses are listed in supplemental Table 2. MPN was present in 2 of 32 (6.3%) CALR positives and in 14 of 613 (2.3%) JAK2 V617F positives. Among mutation positives with MPN (n = 16), PV (n = 10) was the most frequent subdiagnosis (63%). The odds ratio for a diagnosis of MPN per percentage allele burden was 1.14 (95% CI, 1.09-1.18; P = 1.6 × 10−10).
Blood tests
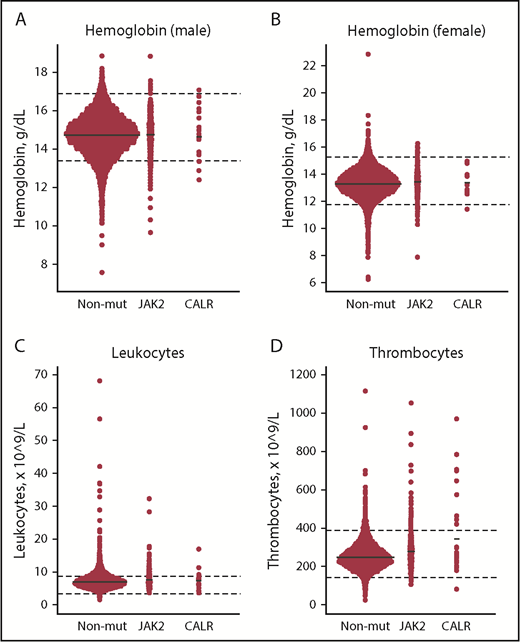
Compared with JAK2 V617F, CALR positives had significantly higher allele burden (mean [standard error (SE)], 7.5% [2.2%] vs 2.1% [0.34%]) and thrombocyte counts (Table 2, A vs B). Compared with nonmutated participants, CALR-positive participants also had higher thrombocyte counts (Table 2, B vs C), and JAK2 V617F positives had higher hemoglobin, leukocyte, and thrombocyte counts, whereas ferritin, high-sensitivity C-reactive protein (hsCRP), and cholesterol levels were lower (Table 2, A vs C). CALR positives had lower eGFR compared with both JAK2 V617–positive (A vs B) and nonmutated participants (B vs C). Key blood cell counts are depicted in Figure 2.
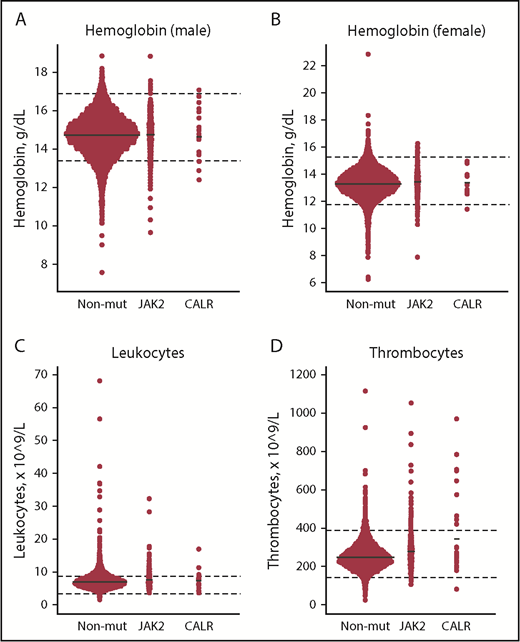
Dot plots of key blood cell counts for the nonmutated, JAK2 V617F–positive, and CALR-positive patients. (A-B) Hemoglobin (male/female). (C) Leukocytes. (D) Thrombocytes. Solid black lines represent means. Dashed black lines represent lower and upper limit ranges. JAK2, JAK2 V617F–positive individuals; Non-mut, nonmutated individuals.
Dot plots of key blood cell counts for the nonmutated, JAK2 V617F–positive, and CALR-positive patients. (A-B) Hemoglobin (male/female). (C) Leukocytes. (D) Thrombocytes. Solid black lines represent means. Dashed black lines represent lower and upper limit ranges. JAK2, JAK2 V617F–positive individuals; Non-mut, nonmutated individuals.
Compared with JAK2 V617F positives with allele burden <1%, JAK2 V617F positives with allele burden ≥1% had higher hematocrit, leukocyte, neutrophil, and thrombocyte counts and lower cholesterol levels (Table 3, A vs B). Both JAK2 V617F groups had significantly higher leukocyte, neutrophil, and thrombocyte counts and lower ferritin levels compared with nonmutated participants (Table 3, A vs C and B vs C). Also, JAK2 V617F positives with allele burden ≥1% had lower cholesterol levels, and JAK2 V617F positives with allele burden <1% had lower hsCRP compared with the nonmutated participants.
Compared with mutation positives without MPN (non-MPNs), mutation positives with MPN had a higher allele burden (mean [SE], 35% [8.1%] vs 1.6% [0.20%]) and hsCRP (Table 4, A vs B). Also, leukocyte and thrombocyte counts and alkaline phosphatase levels were higher, whereas hemoglobin, ferritin, cholesterol, and eGFR were lower, compared with both mutation-positive non-MPNs (A vs B) and nonmutated (B vs C). Mutation-positive non-MPNs had higher hemoglobin, leukocytes, neutrophils, and thrombocytes and lower ferritin, hsCRP, and cholesterol compared with nonmutated (A vs C).
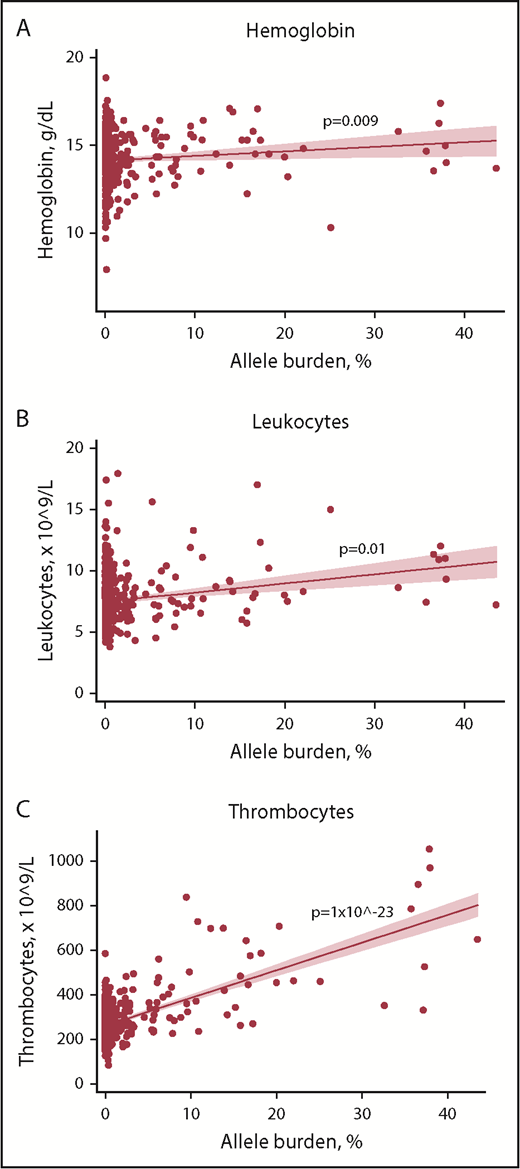
In mutation-positive non-MPNs, positive associations between blood cell counts and allele burden were detected for hemoglobin, leukocytes, and thrombocytes (Figure 3). Individuals with current MPN were not included in this analysis due to an anticipated altered blood cell profile by cytoreductive therapy.
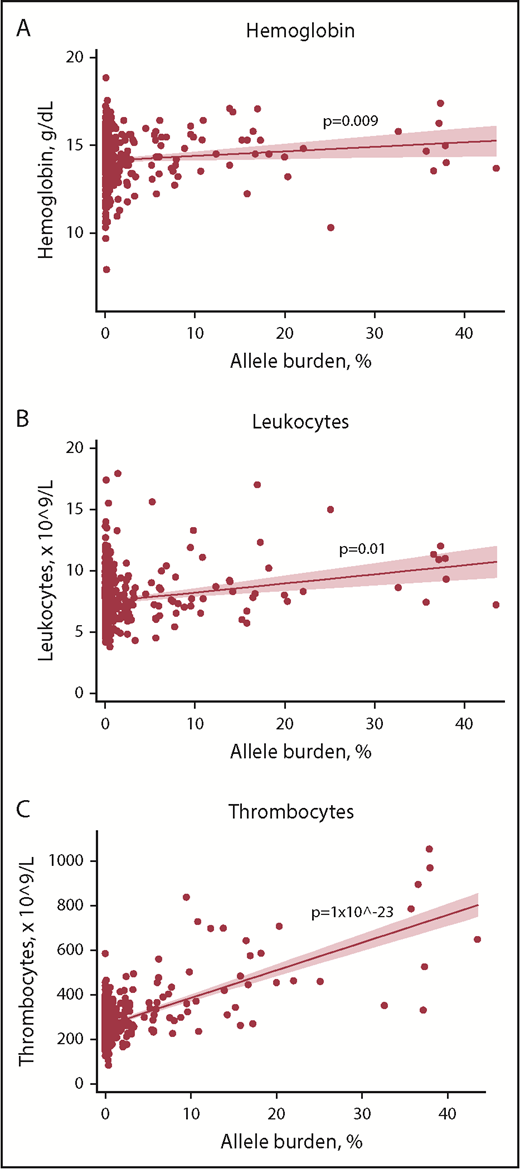
Scatterplots with regression line of mutation-positive non-MPNs with allele burden vs key blood cell counts. (A) Hemoglobin. (B) Leukocytes. (C) Thrombocytes. Solid red line represents the regression line (unadjusted). Red shading represents the 95% CI.
Scatterplots with regression line of mutation-positive non-MPNs with allele burden vs key blood cell counts. (A) Hemoglobin. (B) Leukocytes. (C) Thrombocytes. Solid red line represents the regression line (unadjusted). Red shading represents the 95% CI.
Additional blood tests for all groups are presented in supplemental Tables 3-5.
Of the 629 mutation-positive non-MPNs, 262 (42%) had elevated blood cell counts above upper normal range (Table 5). Of these, 66%, 20%, 9%, and 5% had elevation of 1, 2, 3, or ≥4 blood cells, respectively, and the most commonly involved cell types were white blood cells. Median allele burden was 0.14% (interquartile range, 0.049% to 0.84%), and the mutational distribution was 13 CALR- and 249 JAK2 V617F–positive participants.
Furthermore, 80 (13%) of the mutation-positive non-MPNs presented blood cell counts in accordance with the current WHO diagnostic criteria of ET, PV, prefibrotic myelofibrosis, or PMF.3
Thrombosis
The odds ratio (model 1) for prevalent venous thromboembolism (deep vein thrombosis and/or pulmonary embolism) was 2.8 (95% CI, 1.1-7.0; P = .02) for JAK2 V617F–positive non-MPNs with allele burden ≥1% and 6.0 (95% CI, 1.3-28; P = .04) for mutation-positive MPNs compared with nonmutated individuals (Table 6). Additional adjustment for blood cell counts (model 2) slightly changed the odds ratio, but results remained significant, suggesting mediation by (although not limited to) blood cell counts. The odds ratio (model 1) for prevalent ischemic cerebrovascular disease was 4.6 (95% CI, 1.2-18; P = .03) in mutation-positive MPNs; additional adjustment for blood cell counts (model 2) decreased the odds ratio, which also became nonsignificant, suggesting mediation primarily by blood cell counts.
Lifestyle factors
Lifestyle factors are presented for all mutation positives and the total nonmutated background population for completion (Table 7). To avoid possible reverse causation, we also analyzed lifestyle factors in a subpopulation with no smoking-related diseases; more current smokers and heavy smokers (≥10 pack-years) were found among mutation positives as well as a significant positive trend in smoking exposure for mutation positives. The mutation positives from the subpopulation also had a significantly higher alcohol consumption than nonmutated participants.
Among mutation positives, the frequency of current smokers also increased by number of elevated blood cell counts (P = 6.1 × 10−5) (supplemental Table 6). In the total population, the frequency of current heavy smokers was higher in those with elevated blood cell counts both with (25%) and without mutation (24%) and among MPNs (19%) compared with smokers without elevated blood cell counts irrespective of mutational status (9.7% and 9.5%) (P = 3.2 × 10−157) (supplemental Table 7).
Discussion
In a population-based screening of 19 958 adult citizens by highly sensitive ddPCR, we found a novel CALR population prevalence of 0.16% with mean allele burden of 7.5% and a JAK2 V617F population prevalence of 3.1% with a mean allele burden of 2.1%. The population-based JAK2 V617F:CALR ratio was 19:1. These are novel findings. Increasing age, smoking, and alcohol consumption were risk factors for these mutations. Risk of MPN increased 14% per percentage allele burden. Mutation positives displayed higher blood cell counts compared with nonmutated participants, and 42% of mutation positives without MPN presented elevation in ≥1 blood cell counts.
CALR
We are the first to report a CALR prevalence in the general population and to substantiate presence of CALR in non-MPNs.15 Compared with the population prevalence of JAK2 V617F, CALR is rare, which could be explained by (1) the structural biology of the mutations, meaning that larger insertions/deletions (CALR) ought to be less prone to happen by chance than a point mutation (JAK2 V617F); or (2) the more pronounced immunogenicity of CALR compared with JAK2 V617F, which in theory means that the adaptive immune system might be able to control or even clear CALR.31 Also, the slight difference in sensitivity of the CALR assay vs JAK2 V617F could account for part of the prevalence difference.
To compare our JAK2 V617F:CALR ratio from the general population with the corresponding prevalence ratio within MPN, we calculated a rough estimate of 5.6:1 for the latter based on national MPN prevalence numbers from Brochmann et al32 and the mutational distribution within MPN from Nangalia et al.2 The marked difference between our population-based JAK2 V617F:CALR ratio and the MPN-based JAK2 V617F:CALR ratio suggests that a higher percentage of the CALR positives in the general population evolves into MPN compared with JAK2 V617F positives. This is supported by our MPN data, where ∼3 times as many of the CALR positives have a MPN diagnosis compared with the frequency within JAK2 V617F, and that the mean CALR allele burden is significantly higher than for JAK2 V617F. Our observations are in line with the striking absence of reports of low CALR allele burdens in newly diagnosed/untreated MPN patients, suggesting that, when present, the mutation quickly takes dominance of the stem cell compartment and consistently entails a MPN phenotype, as also suggested by others.33 A CALR (type 1) mouse model is also indicative of this assumption,34 as well as patient data showing similar high levels of CALR allele burden in progenitor and hematopoietic stem cells when comparing early MPN (ET) with late-stage disease (PMF).35 In contrast, the JAK2 V617F allele burden significantly differs between MPN subdiagnoses,35 and JAK2 V617F has been detected in healthy volunteers, in general presenting very low allele burdens.4,5 Low JAK2 V617F allele burdens <1% have also been described in newly diagnosed MPN patients,36,37 and the low allele burdens can be stable for years in a fraction of patients.38 Accordingly, we find our novel results regarding CALR positivity and low allele burdens in non-MPN individuals intriguing. Do these individuals constitute a healthy entity with stable/diminishing CALR allele burdens, or will they eventually experience increasing allele burdens with emergence of overt MPN? If the latter is the case, we might need to change our approach to “healthy” individuals with low JAK2 V617F allele burdens correspondingly and consider them as (pre)MPNs in the very early stage of the biological continuum CHIP-ET-PV-PMF; a viewpoint also supported by the high hazard ratios of 11 and 12.9 for hematological malignancies in individuals with detectable mutation found in the above-mentioned CHIP studies26,27 and by our data showing significant association with MPN by increasing allele burden. According to an estimate from a mathematical model on MPN development, the time span from the first mutational hit to overt MPN (represented by JAK2 V617F allele burden of 7%) is 24 years,39 which underscores the relevance of an early diagnosis as this long prediagnosis phase allows preventive and interventional actions.
JAK2 V617F
The JAK2 V617F prevalence of 3.1% in our general population study is 3 to 30 times higher than in earlier reports,25-29 whereas the MPN prevalence was found much lower with only 14 of 613 (2.3%) JAK2 V617F positives. Nielsen et al found a JAK2 V617F prevalence of 0.1% of which 19% had MPN (13 of 68) among 49 488 citizens in a Danish population–based screening.25 Genovese et al found JAK2 V617F mutations in 0.19% of which 29% (7 of 24) had MPN among 12 380 cases (bipolar disorders or schizophrenia) and controls.27 Jaiswal et al found a JAK2 V617F prevalence of 0.18% among 17 182 individuals mainly from cohorts of diabetes cases and controls.26 McKerrell et al found a JAK2 V617F prevalence of 0.61% among 4067 blood donors and participants from the general population.28 Xu et al found a JAK2 V617F prevalence of 0.94% among 3935 random blood samples from a Chinese hospital.29 Detailed information on MPN diagnoses was not available from Jaiswal et al,26 McKerrell et al,28 or Xu et al.29 The discrepancies between the studies can likely be explained by the difference in the sensitivity of the assays (supplemental Table 1). Thus, in studies using methods with a high sensitivity, like ours, more participants having very low allele burdens will be detected (supplemental Table 1), whereas in studies using methods with lower sensitivity, the detection limit is less sensitive leading to a higher MPN prevalence.
MPN phenotype
Our data reveal that all mutation positives irrespective of subgrouping present a more hyperproliferative blood cell count profile compared with the nonmutated background. The fact that two-fifths of the mutation-positive non-MPNs have elevation of ≥1 blood cell count and >10% present elevated blood cell counts meeting the current MPN diagnostic criteria underscore the resemblance with the MPN phenotype suggesting that some mutated participants remain undiagnosed for MPN in the population. If just half of the 80 high-risk individuals with elevated blood cell counts meeting diagnostic criteria turn out to be true MPN patients, the MPN prevalence in our study population increases 350%. Extrapolated to nationwide numbers, >14 000 individuals in Denmark may suffer from MPN, of which almost 10 000 would be unrecognized at time of GESUS based on the prevalence of Philadelphia chromosome–negative MPNs in Denmark in 2013 reported by Brochmann et al.32 This is in line with the yearlong prediagnosis MPN phase, where patients may have elevated blood cell counts elicited by thrombotic events and/or concurrent chronic inflammatory conditions40,41 but that have either been interpreted as “reactive” or remained undetected. The majority of these individuals with elevated blood cell counts had allele burdens well below 1%, and combined with the positive association detected between allele burden and blood cell counts in mutation-positive non-MPNs, these findings add to the unresolved issue on how to interpret low JAK2 V617F allele burdens in relation to (early) MPN diagnostics when all conventional diagnostic criteria are not (yet) met. In our opinion, these individuals should be considered as high risk for progressing to overt MPN, and studies on this highly important issue are urgently needed.
The odds for prevalent venous thromboembolism were higher in MPNs compared with nonmutated individuals, which is in line with previous studies.40,41 As a novel finding, we detected a similar risk in JAK2 V617F–positive non-MPNs (representing CHIP), which is in line with a recent study of 10 000 citizens showing significantly higher thrombotic rates with CHIP (especially JAK2 V617F) than with non-CHIP.42 In this context it is intriguing to consider enhanced inflammation (eg, smoking18,19 or comorbidities such as inflammatory bowel disease or rheumatic diseases41,43 ), occurrence of comutations (such as Ten–Eleven Translocation-244 ), or presence of a particular haplotype (eg, 46/145 ) as promotors for progression from CHIP toward overt MPN.
The decreased iron markers in most of the mutation-positive subgroup are consistent with existing MPN evidence, especially in PV patients, in whom chronic inflammation is an important part of the complex mechanisms of the sustained iron deficiency.46,47 Decreased eGFR in the MPN and CALR subgroups complies with the frequent occurrence of chronic kidney disease reported within MPNs,48 and together with higher alkaline phosphatase, it could also be indicative of a general organ involvement from systemic inflammation present in cancer,49 including MPN50-53 and other myeloid malignancies.53 Systemic inflammation is also an apparent explanation for the elevated level of hsCRP found in our MPNs in accordance with previous findings.54-56 As we argue that hsCRP is associated with chronic inflammation and chronic inflammation is associated with mutation/MPN,57,58 we have no obvious explanation for the lower hsCRP levels within other mutation-positive subgroups compared with the nonmutated background. However, CRP levels are influenced by several health-related, genetic, and lifestyle factors that we have not adjusted for.59
The lipid profile with low cholesterol values in almost all mutation-positive subgroups is in accordance with observations from MPN patients60-62 as well as other cancers.63,64 Although the exact mechanisms are not fully understood, cholesterol seems to promote cancer cell growth, thereby inducing a plasma cholesterol depletion, which makes the hypocholesterolemic state a symptom of progressive cancer.65
Lifestyle
Based on earlier reports of significant associations between smoking and MPN,18-21 we hypothesized that a similar association exists between smoking and mutation,16 as smoking is a strong inflammatory stimulus66 and the presence of a driver mutation is considered a “precursor state” for overt MPN. We are concerned about the detected association of current smoking and the presence of mutation as well as the increasing number of elevated blood cell counts because of the implication of future MPN occurrence. Our data suggest that the association is characterized by ongoing considerable smoking exposure, which indicates that smoking cessation matters, as also reported within other myeloid malignancies.23 Acting as an inflammatory driver, we speculate whether sustained smoking will induce a “right shift” of individuals over time among the current heavy smokers in supplemental Table 4 so that more will become mutation positive and experience elevated blood cell counts, ultimately leading to overt MPN. Thus, we find this an additional strong argument for early mutational detection in that proactive smoking cessation might diminish further evolvement toward MPN.
Alcohol is yet another immune modulator, though less pronounced than smoking.67 Alcohol consumption in relation to MPN and myelodysplasia/MPN has been assessed in a few studies, with no association detected.20,21 However, alcohol has been suggested to have a protective effect on the risk of lymphoid malignancies.68,69 This inverse relationship was not present in our data, and we interpret our results as indicative of a nonbeneficial and perhaps harmful effect of alcohol on mutational status.
Strengths and limitations
The strengths of this study are the population-based unselected cohort of adult citizens at a total number large enough to detect even rare events and associations and with confirmation of MPN diagnosis by electronic medical records. Also, the use of a highly sensitive method brings novel information to the area. Limitations include the cross-sectional design from which conclusions on causality cannot be drawn, but in future follow-up studies, we will be able to predict the risk of thromboembolism, especially for those with CHIP. Another limitation is the possible reduced sensitivity in part of the analyses described in supplemental Methods, as this may have bypassed some true low burden mutations but we estimate the prevalence of JAK2 V617F and CALR to be representative, and we do not consider it to have a significant impact on the results presented in the paper. With regard to CALR, the fact that we have only tested for type 1 and 2 mutations obviously implies that the true total CALR prevalence may be higher assuming that type 1–like, type 2–like, and type 3 CALR are present in the background population as well.
Conclusion
In summary, we are the first to report a CALR prevalence (type 1 and 2) in the general population. We also find a JAK2 V617F population prevalence much higher than previously detected. When comparing mutation-positive citizens with the nonmutated background population, we discover several significant differences in blood test results, suggesting that the presence of a driver mutation is associated with a distinct paraclinical profile that mimics the MPN phenotype, also present in individuals with allele burdens <1% and no MPN diagnosis registered. As 42% of mutation-positive non-MPNs have elevated blood cell counts, we have concerns that many of these individuals have an undiagnosed MPN. We therefore suggest that individuals with a driver mutation, even at a very low mutant allele burden, are considered MPN-risk patients. In this context, our finding of elevated odds for prevalent venous thromboembolism also in JAK2 V617F–positive non-MPNs is supportive of more regular screening for JAK2 V617F in “target populations” at risk of MPN. Furthermore, we argue that a focus on smoking cessation and possibly minimizing alcohol consumption can be of considerable importance in mutation-positive individuals, including those with no comorbidity, as changes in these lifestyle factors might diminish further progression towards MPN by reducing the driving inflammatory stimulus.
For original data, please contact christina.ellervik@childrens.harvard.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the Region Zealand Research Foundation, Manufacturer Einar Willumsens Memorial Foundation, Anders Hasselbalch’s Foundation Fighting Leukemia, Carpenter Joergen Holm and Wife Elisa F. Hansen’s Memorial Foundation, Else and Mogens Wedell-Wedellborgs Foundation, the Hoejmosegaard Scholarship, Eva and Henry Fraenkels Memorial Foundation, Dagmar Marshalls Foundation, Candys Foundation, A.V. Lykfeldt and Wife’s Scholarship, and Aase and Ejnar Danielsen’s Foundation. GESUS was funded by the Region Zealand Research Foundation, Naestved Hospital Foundation, Naestved Municipality, Johan and Lise Boserup Foundation, TrygFonden, Johannes Fog's Foundation, Region Zealand, Naestved Hospital, The National Board of Health, and the Local Government Denmark Foundation.
S.C. is a PhD candidate at the University of Copenhagen, and this work is submitted in partial fulfillment of the requirement for a PhD.
The sponsors are public or nonprofit organizations that support science in general. They had no role in gathering, analyzing, or interpreting the data.
Authorship
Contribution: H.C.H., C.E., V.S., L.K., and S.C. designed the study; L.K. and N.P. designed the molecular assays used; N.P. performed the analyses; S.C. analyzed results and made tables and figures; and S.C. wrote the paper, with all coauthors substantially contributing to revision and improvement.
Conflict-of-interest disclosure: H.C.H. received a research grant from Novartis. N.P. has received research funding, honorarium, and a travel grant from Novartis; is on the advisory board at Novartis and Qiagen; has received honoraria from Roche and InCyte; and has received research funding from InCyte. The remaining authors declare no competing financial interests.
Correspondence: Sabrina Cordua, Department of Hematology, Zealand University Hospital, Sygehusvej 10, 4000 Roskilde, Denmark; e-mail: scordua@hotmail.com.