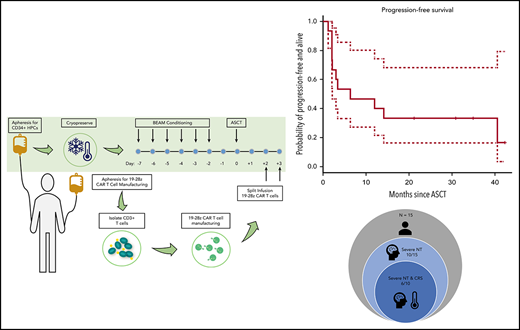
Key Points
19-28z CAR T cells following HDT-ASCT resulted in an incidence of severe neurotoxicity of 67%.
19-28z CAR T grafts with increased effector immunophenotypes trended toward protection from POD following HDT-ASCT.

Abstract
High-dose chemotherapy and autologous stem cell transplantation (HDT-ASCT) is the standard of care for relapsed or primary refractory (rel/ref) chemorefractory diffuse large B-cell lymphoma. Only 50% of patients are cured with this approach. We investigated safety and efficacy of CD19-specific chimeric antigen receptor (CAR) T cells administered following HDT-ASCT. Eligibility for this study includes poor-risk rel/ref aggressive B-cell non-Hodgkin lymphoma chemosensitive to salvage therapy with: (1) positron emission tomography–positive disease or (2) bone marrow involvement. Patients underwent standard HDT-ASCT followed by 19-28z CAR T cells on days +2 and +3. Of 15 subjects treated on study, dose-limiting toxicity was observed at both dose levels (5 × 106 and 1 × 107 19-28z CAR T per kilogram). Ten of 15 subjects experienced CAR T-cell–induced neurotoxicity and/or cytokine release syndrome (CRS), which were associated with greater CAR T-cell persistence (P = .05) but not peak CAR T-cell expansion. Serum interferon-γ elevation (P < .001) and possibly interleukin-10 (P = .07) were associated with toxicity. The 2-year progression-free survival (PFS) is 30% (95% confidence interval, 20% to 70%). Subjects given decreased naive-like (CD45RA+CCR7+) CD4+ and CD8+ CAR T cells experienced superior PFS (P = .02 and .04, respectively). There was no association between CAR T-cell peak expansion, persistence, or cytokine changes and PFS. 19-28z CAR T cells following HDT-ASCT were associated with a high incidence of reversible neurotoxicity and CRS. Following HDT-ASCT, effector CD4+ and CD8+ immunophenotypes may improve disease control. This trial was registered at www.clinicaltrials.gov as #NCT01840566.
Introduction
High-dose chemotherapy (HDT) followed by autologous stem cell transplantation (ASCT; HDT-ASCT) is the established standard of care for patients with relapsed or primary refractory (rel/ref) diffuse large B-cell lymphoma (DLBCL).1 Unfortunately, long-term overall survival (OS) and progression-free survival (PFS) is ∼50% following HDT-ASCT, despite investigational conditioning2 or maintenance3 interventions. Functional imaging response to salvage chemotherapy by positron emission tomography using 18F-deoxyglucose (FDG-PET)4-7 and bone marrow involvement of B-cell non-Hodgkin lymphoma (B-NHL)8 are significant prognostic factors for patients proceeding to HDT-ASCT. Allogeneic hematopoietic cell transplantation has been used to overcome chemoresistance but is compromised by risk of treatment-related mortality secondary to graft-versus-host disease and infectious complications.
Recently, chimeric antigen receptor (CAR)-modified T cells directed against CD19 (CAR19) have demonstrated encouraging activity against a variety of B-cell malignancies.9-15 National Institutes of Health (NIH) investigators previously demonstrated enhanced efficacy of adoptive cell therapy for melanoma by intensifying lymphodepleting conditioning resulting in increasing levels of lymphoproliferative cytokines such as interleukin 7 (IL-7) and IL-15.16 Additionally, in animal models, the infusion of hematopoietic stem cells independently promotes the expansion and function of antitumor T cells.17 Herein, we report a phase 1 study of autologous second-generation CD19 CAR T cells incorporating the signaling domain of the T-cell CD3ζ chain and costimulatory CD28 domain (19-28z CAR)18 with the intention of exploiting myeloablative chemotherapy with lympholytic agents for lymphodepletion following HDT-ASCT. We report here on the safety, tolerability, and efficacy of 19-28z CAR T-cell infusion in patients with rel/ref DLBCL following HDT-ASCT, together with correlative cytokine and immunophenotypic analyses.
Methods
Trial design and oversight
We conducted a phase 1 clinical study of 19-28z CAR T cells in adult patients following HDT-ASCT with chemosensitive poor-risk rel/ref aggressive B-NHL defined by FDG-PET+ disease following salvage chemotherapy (Deauville 4 on the 5-point Deauville scale19 ) and/or bone marrow involvement of rel/ref B-NHL (NCT01840566). Dose-limiting toxicities (DLTs) on the phase 1 study are defined in supplemental Table 1 (available on the Blood Web site). The protocol was approved by the Memorial Sloan Kettering Cancer Center (MSKCC) institutional review board and was conducted according to the Declaration of Helsinki principles. Subjects were enrolled upon eligibility criteria being met after at least 2 cycles of salvage chemotherapy.
Following consent, subjects underwent 2 separate apheresis procedures including: granulocyte colony-stimulating factor (G-CSF)-primed hematopoietic progenitor cell collection and apheresis for CD3+ CAR T production. The median number of days between the cryopreservation of the 19-28z CAR product and the ASCT was 14 days (range, 7-78 days). From the apheresis product for CAR T generation, CD3+ cells were selected and activated using Dynabeads CD3/CD28 and transduced with SFG-19-28z retroviral vector as previously described.13,20,21 Transduced cells were expanded per dose requirement, formulated, and released as previously described.17,20,21 Subjects were subsequently admitted to MSKCC and received standard carmustine, etoposide, cytarabine, and melphalan (BEAM) conditioning prior to infusion of a minimum 2 × 106 CD34+ stem cells per kilogram. Pegfilgrastim, 6 μg subcutaneously 1 time, was administered 1 day following stem cell infusion. 19-28z CAR T cells were infused days +2 (two-thirds of the dose) and +3 (one-third of the dose) in split dosing per the phase 1 design (Figure 1).
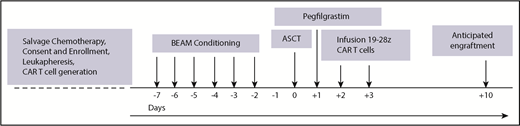
Study treatment schema. BEAM, carmustine, etoposide, cytarabine, and melphalan.
Study treatment schema. BEAM, carmustine, etoposide, cytarabine, and melphalan.
Toxicity assessment
Cytokine release syndrome (CRS) was graded according to the American Society for Blood and Marrow Transplantation (ASBMT) Consensus Criteria.22 Neurotoxicity was assessed according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) v 4.03. Severe neurotoxicity was defined as any grade seizure or ≥grade 3 nervous system and psychiatric disorders including, but not limited to: encephalopathy, depressed level of consciousness, lethargy, somnolence, confusion, dysphasia, dysarthria, tremor, delirium, and/or hallucinations.
Staging and response criteria
Chemosensitivity appropriate for HDT-ASCT was assessed per standard computed tomography criteria for B-NHL.23 High-risk patients were eligible for study if FDG-PET was deemed positive as a Deauville score 4 (FDG more than background liver uptake) per Deauville criteria.19 A Deauville 5 FDG-PET scan constituted progression of disease (POD) following study treatment. All patients underwent standard bone marrow biopsy procedures prior to hematopoietic progenitor cell collection and/or at the time of restaging of rel/ref disease prior to pre–HDT-ASCT salvage chemotherapy.
Immunophenotype of CAR T cells
End of production CAR T cells were stained with biotinylated F(ab) goat anti-mouse (Jackson ImmunoResearch Laboratories) to detect expression of 19-28z. After incubation with 2% mouse serum to prevent nonspecific staining, a pool of antibody including CD3 peridinin chlorophyll protein–Cy5.5 (Becton Dickinson), CD8 phycoerythrin-Cy7 (BD Biosciences), CD45RA allophycocyanin (Invitrogen), and CCR7 fluorescein isothiocyanate (R&D Systems) was added. Stained cells were acquired on an LSRII flow cytometer (BD Biosciences). Analysis was performed using FlowJo software. Expression of CD45RA and CCR7 was assessed on transduced CD8 T cells (CD3+, CAR+, CD8+) and transduced CD4 T cells (CD3+, CAR+, CD8−).
Assessment of 19-28z CAR T-cell expansion by quantitative PCR
Presence of 19-28z CAR T-cell was detected by polymerase chain reaction (PCR) from peripheral blood as previously described.21 EDTA blood was collected for monitoring the detection of CAR T cells before infusion and daily during the first week after infusion, weekly for 6 weeks, monthly thereafter until 12 months. The vector copy number was reported per microliter of blood to study expansion and persistence of CAR T cells after infusion. Persistence is defined as the number of days after infusion that the peripheral blood was last reported positive by PCR.
Assessment of cytokines released
Peripheral blood serum cytokine levels were determined using the Milliplex 38-cytokine panel on a Luminex FlexMap3D instrument with Xponent 4.2 software, and determined at the day of ASCT, at 5 days, and 10 days after ASCT (supplemental Figure 1).
Statistical methods
Continuous variables were summarized by median and range. Categorical variables were summarized by frequency and percentage. The 95% confidence interval (CI) for proportions was calculated based on exact binomial distribution. The within-subject change in cytokine levels was calculated by ratios. Because the measured cytokine level was subject to a lower detection limit (LDL), the ratio was considered right censored if the denominator was below LDL and left censored if the numerator was below LDL. The record was excluded if both denominator and numerator were below LDL. Due to the censoring, the median ratio was computed by Kaplan-Meier method and the 95% CI was computed using the Greenwood formulas. The median ratio was compared between groups using the log-rank test and correlated with continuous variables using Cox regression. Continuous variables without censoring were compared between groups using the Wilcoxon rank-sum test and correlated with other continuous variables using the Spearman correlation coefficient. Categorical variables were compared using the Fisher exact test. PFS was calculated from time of CAR T-cell administration to POD or death. Because there is no censoring, the PFS at a given time point was computed as a simple proportion. All tests were 2-sided at the 0.05 significance level. Statistical analyses were conducted in SAS 9.4 (SAS Institute, Cary, NC) and R 3.3.2.24
Results
Subjects and control cohort
Between June 2013 and September of 2015, 17 adult subjects with aggressive histology rel/ref B-NHL were enrolled and 15 subjects were treated on study with 19-28z CAR T cells. Of the 2 patients who did not proceed to study treatment: 1 developed sepsis followed by POD and the other experienced POD prior to planned HDT-ASCT on study. The median age of the subjects was 61 years and 7 of 15 subjects (47%) had 3 or more prior lines of therapy with 1 subject having received CD19-targeted CAR T cells twice previously at 2 different institutions outside of MSKCC. Three of the 15 subjects had bone marrow involvement of B-NHL at the time of rel/ref disease. One subject, with rel/ref Burkitt lymphoma, had persistent minimal residual disease positivity in the bone marrow at the time of HDT-ASCT on study. Fourteen of 15 subjects were FDG-PET+ at the time of HDT-ASCT. Subjects’ characteristics are outlined in Table 1.
19-28z CAR T-cell manufacturing
The eligibility criteria for the study did not exclude patients based on absolute lymphocyte count. Median absolute lymphocyte count at the time of leukapheresis was 400/µL (range, 200-1100). Apheresis products were analyzed for absolute T-cell counts and revealed variable CD4+-to-CD8+ T-cell ratios that increased significantly during the manufacturing process in both transduced and untransduced T cells (supplemental Figure 2). γ-retroviral 19-28z CAR gene transfer was robust with a mean CAR transduction efficiency of 42.2% (range, 15% to 70%). In this heavily pretreated patient population, the protocol-specified CAR T-cell dose (5 × 106 to 1 × 107 19-28z CAR T cells per kilogram) was successfully produced for all subjects recently completing salvage chemotherapy.
Safety and tolerability after CAR T-cell infusion
Dose level #1 (DL#1) on the 3 + 3 design was 5 × 106 19-28z CAR T cells per kilogram. Following no observed DLTs in the first 3 subjects at DL#1, 1 subject was treated at DL# 2 (1 × 107 19-28z CAR T cells per kilogram) and experienced the only case of severe CRS on study (grade 4). At this point, given the severe toxicity observed at DL#2, it was the decision of the investigators to expand enrollment at DL#1. One additional subject experienced a DLT with grade 5 pulmonary mucormycosis not attributed to 19-28z CAR T-cell infusion. The most common unique toxicity, outside of expectant cytopenias from HDT, including 13 of 15 patients with uncomplicated febrile neutropenia, was 10 of 15 subjects (67%) experiencing grade 3-4 neurotoxicity and/or seizure in the study subjects. The median time to onset of neurotoxicity was 5 days (range, 1-6 days) post 19-28z CAR T infusion with resolution with a median duration of 9.5 days (range, 2-20 days). Of the 10 subjects who experienced neurotoxicity, 6 also experienced grade 2-4 CRS at a median of 2.5 days (range, 0-10 days) post 19-28z CAR T infusion with a median duration of 8 days (range, 3-12 days). All grade 2 CRS with associated hypotension are grade 3 per Penn criteria.25 No subjects experienced CRS without neurotoxicity (Table 1). Nine of the 10 subjects who experienced severe neurotoxicity received tocilizumab, n = 4 subsequently received dexamethasone and 1 subject was observed. All neurotoxicity and CRS subsequently resolved.
Correlation of toxicities with cytokines and CAR T-cell expansion
Serum cytokines, c-reactive protein (CRP), and 19-28z CAR T-cell expansion and persistence were analyzed for correlation to neurotoxicity and CRS on study. In the subjects who experienced neurotoxicity, a longer persistence of 19-28z CAR T cells (median of 11 days; range, 4-22 days) was observed compared with subjects who did not experience neurotoxicity (median, 4 days; range, 1-8 days; P = .05) (Figure 2A). There was no significant difference in peak expansion of CAR T cells in subjects without neurotoxicity with a median of 141 vector copies per microliter (range, 22-852) compared with a median of 15 vector copies per microliter (range, 1-249) in the subjects who experienced neurotoxicity (P = .16) (Figure 2B; supplemental Figure 3). Serum cytokine changes measured pre-CAR T-cell infusion and 3 days post (5 days post-ASCT) showed that the median fold change in interferon-γ (IFN-γ) was significantly higher in subjects experiencing toxicity (6.2-fold increase) in contrast to those who did not (0.9-fold decrease; P < .001) and a trend toward significant increase in IL-10 in subjects with toxicity (9.1-fold increase) contrasted to those who did not (5.3-fold increase; P = .07) (Figure 3). Other serum cytokine changes were comparable between the 2 subject groups. In subjects who experienced CRS, there was a trend toward a significant increase in IL-6 1 week following CAR T-cell infusion (5.1-fold increase) contrasted to those who did not experience CRS (1.1-fold increase, P = .09). Increase in CRP levels 3 and 7 days after CAR T-cell infusion (5 and 10 days post-ASCT) was similar in subjects who experienced or did not experience toxicity (Figure 3).

Toxicity related to 19-28z CAR T-cell expansion. (A) Persistence in days was longer in subjects experiencing severe toxicity. (B) No significant difference was seen in peak values of vector copy number (VCN) per microliter by quantitative PCR.
Toxicity related to 19-28z CAR T-cell expansion. (A) Persistence in days was longer in subjects experiencing severe toxicity. (B) No significant difference was seen in peak values of vector copy number (VCN) per microliter by quantitative PCR.
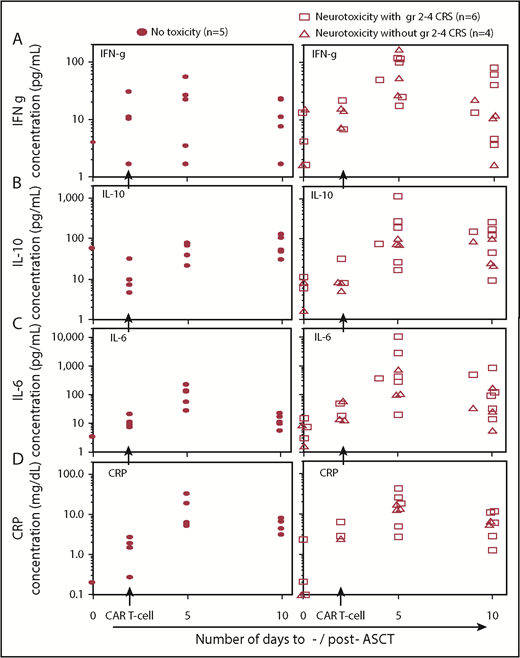
Serum cytokines and CRP levels in subjects developing toxicities. (A) IFN-γ (IFN-g), (B) IL-10, (C) IL-6, and (D) CRP concentrations increase after infusion of CAR T cells (2 days following ASCT) in all patients. Both IFN-γ and IL-10 increased significantly at day 5 post-ASCT in subjects developing a neurotoxicity plus or minus CRS than in subjects without toxicities (P < .001 and P = .07, respectively). There was a trend for significant increase of IL-6 concentration in subjects with CRS than in subjects without CRS. Increases in CRP levels were similar in subjects with and without toxicities.
Serum cytokines and CRP levels in subjects developing toxicities. (A) IFN-γ (IFN-g), (B) IL-10, (C) IL-6, and (D) CRP concentrations increase after infusion of CAR T cells (2 days following ASCT) in all patients. Both IFN-γ and IL-10 increased significantly at day 5 post-ASCT in subjects developing a neurotoxicity plus or minus CRS than in subjects without toxicities (P < .001 and P = .07, respectively). There was a trend for significant increase of IL-6 concentration in subjects with CRS than in subjects without CRS. Increases in CRP levels were similar in subjects with and without toxicities.
PFS
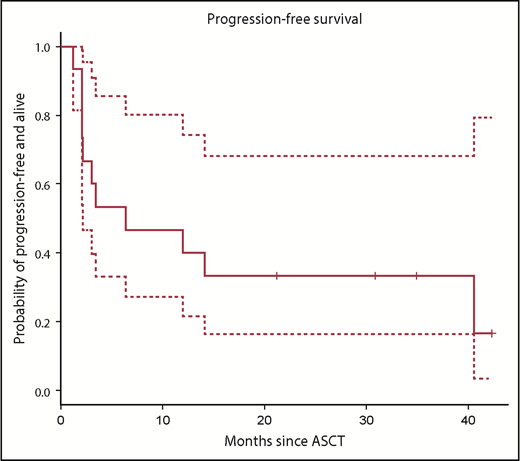
With a median follow-up of 24 months (range, 12-37 months), 4 subjects are alive and progression-free and the PFS at 1 and 2 years post-HDT-ASCT followed by 19-28z CAR T-cell infusion was 40% (95% CI, 20% to 70%) and 30% (95% CI, 20% to 70%), respectively (Figure 4). One subject experienced a late, retrograde transformation of follicular lymphoma to a single-port site of disease and has since undergone involved-site radiotherapy into a continuous complete response (CR). 19-28z CAR T-cell expansion was similar in subjects who remain alive and progression-free with a median peak of 27 vector copies per microliter (range, 9-141) and in subjects who experienced POD and/or death with a median peak of 22 vector copies per microliter (range, 0.1-852) (P = .10; Figure 5A). There was no difference in persistence of 19-28z CAR T cells between subjects who experienced progression and/or death event (median, 8 days; range, 1-22 days) and those subjects who remain alive and progression-free (median, 8 days; range, 6-22 days) (Figure 5B). There was no correlation observed between serum cytokine changes post 19-28z CAR T infusion and likelihood of PFS. Of the 10 subjects who experienced POD, 9 were rebiopsied and 2 were found to be CD19− on rebiopsy at the time of progression (representative case, Figure 6).
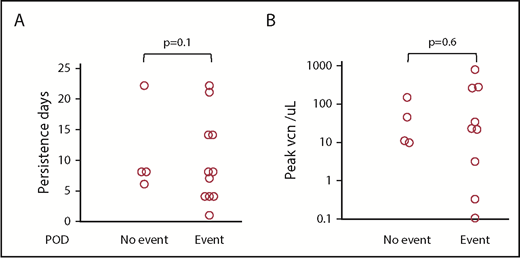
Events (progression or death) related to 19-28z CAR T-cell expansion. (A) Persistence in days and (B) peak according to vector copy number per microliter peripheral blood by quantitative PCR.
Events (progression or death) related to 19-28z CAR T-cell expansion. (A) Persistence in days and (B) peak according to vector copy number per microliter peripheral blood by quantitative PCR.
![Figure 6. Representative progression of DLBCL negative for CD19 surface expression. (A) Core biopsy shows involvement by DLBCL (hematoxylin-and-eosin [H&E] stain). (B) By immunohistochemistry, the neoplastic B cell strongly expresses B-cell lineage marker CD20 with cell-surface pattern (CD20 immunostain). (C) In contrast, CD19 is only weakly expressed with an exclusively cytoplasmic pattern without surface expression (CD19 immunostain). (A-C) Original magnification ×400.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/7/10.1182_blood.2018883421/3/m_blood883421f6.png?Expires=1767861086&Signature=TOQEqkeKgkxEQhbe7plzxe6QT0Vh2gTbF504MjHWek9bQ1qUmt02od-gDIlk~RD1ovUC-e1GrqgUPJl8JGGbeGBZgg3NiIbFvEO6P0Thv131UiMN9fO1uz6HlEsyedO7J-qyWt2tp2A6hc2w3gLrvl~XIBjHEgwEfwQclp2xqDZTm-2r2uwiJl0oYSQrhlvFxSoz7SB-LTpE3qQZVozLLwS4vizzDU7wm0tGacgDwqiUhw~CxXG2scC7qIXSjx-lYVDAX9JT4r9jWbGwGF94jNMV1H5KDs2SfDJQPf8bVyA~lAc96t-Mz42Rhq1~ZOCscv7s4c-WRcvbU1qXRbzW9w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Representative progression of DLBCL negative for CD19 surface expression. (A) Core biopsy shows involvement by DLBCL (hematoxylin-and-eosin [H&E] stain). (B) By immunohistochemistry, the neoplastic B cell strongly expresses B-cell lineage marker CD20 with cell-surface pattern (CD20 immunostain). (C) In contrast, CD19 is only weakly expressed with an exclusively cytoplasmic pattern without surface expression (CD19 immunostain). (A-C) Original magnification ×400.
Representative progression of DLBCL negative for CD19 surface expression. (A) Core biopsy shows involvement by DLBCL (hematoxylin-and-eosin [H&E] stain). (B) By immunohistochemistry, the neoplastic B cell strongly expresses B-cell lineage marker CD20 with cell-surface pattern (CD20 immunostain). (C) In contrast, CD19 is only weakly expressed with an exclusively cytoplasmic pattern without surface expression (CD19 immunostain). (A-C) Original magnification ×400.
19-28z CAR T immunophenotype and correlation to PFS and toxicity
We sought to correlate T-cell immunophenotype composition of the infused CAR T product with PFS. Subjects given fewer naive-like (CD45RA+CCR7+) CD4+ (P = .02) and CD8+ (P = .04) 19-28z CAR T cells were more likely to experience superior PFS (Figure 7A). Although CD4+ central memory (CD45RA−CCR7+) CAR T cells did not correlate with PFS (P = .34), there was a trend toward increased CD8+ central memory CAR T cells to improve PFS (P = .11). Neither CD8+ or CD4+ effector memory (CD45RA−CCR7−) nor effector (CD45RA+CCR7−) 19-28z CAR T cells correlated to PFS. The CD4+-to-CD8+ ratio of the infused cell product (range, 0.34-13.4) had no impact on PFS (P = 1.00). There was no correlation between 19-28z CAR T-cell immunophenotype and toxicity postinfusion (Figure 7B).
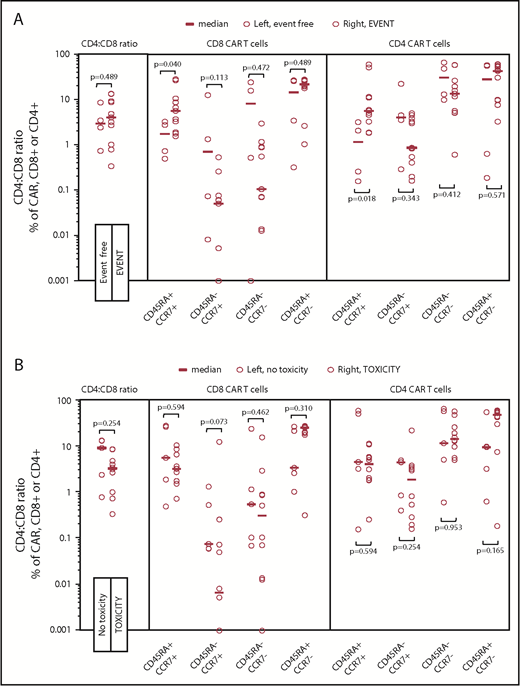
Immunophenotype of 19-28z CAR T-cell products infused and relationship to events (progression or death) and toxicity. Open circles represent data points for each subjects’ graft products’ immunophenotype proportion. (A) Events. Subjects whose infused CAR T cells were composed of a greater percentage of naive-like (CD45RA+, CCR7+) cells were more likely to experience an event for both CD4+ (P = .02) and CD8+ (P = .04) CAR+ subsets. No significant difference was seen in other immunophenotypic subsets: central memory (CD45RA−, CCR7+), effector memory (CD45RA−, CCR7−), and effector (CD45RA+, CCR7−) T cells. (B) Toxicity. No significant differences were seen in any immunophenotypic subsets of the 19-28z CAR T graft product between subjects who did or did not experience toxicity.
Immunophenotype of 19-28z CAR T-cell products infused and relationship to events (progression or death) and toxicity. Open circles represent data points for each subjects’ graft products’ immunophenotype proportion. (A) Events. Subjects whose infused CAR T cells were composed of a greater percentage of naive-like (CD45RA+, CCR7+) cells were more likely to experience an event for both CD4+ (P = .02) and CD8+ (P = .04) CAR+ subsets. No significant difference was seen in other immunophenotypic subsets: central memory (CD45RA−, CCR7+), effector memory (CD45RA−, CCR7−), and effector (CD45RA+, CCR7−) T cells. (B) Toxicity. No significant differences were seen in any immunophenotypic subsets of the 19-28z CAR T graft product between subjects who did or did not experience toxicity.
Discussion
Herein, we report our results of 19-28z CAR-modified T cells in consolidation following HDT-ASCT in poor-risk rel/ref aggressive B-NHL subjects with a 1- and 2-year PFS of 40% and 30%, respectively. In this phase 1 study, high-risk subjects were enrolled as characterized by FDG-PET+ following salvage and with historic 2-year PFS approximating 30% to 50%.4-7 In our recent analysis, we have described FDG-PET response to salvage therapy in patients proceeding to HDT-ASCT as the single dominant prognostic factor beyond other established risk factors for salvage chemotherapy followed by HDT-ASCT per the intention-to-treat Collaborative Trial in Relapsed Aggressive Lymphoma (CORAL) study26 including: age-adjusted International Prognostic Index (IPI) and early relapsed or refractory disease in the postrituximab era.4 We included subjects with bone marrow involvement at the time of rel/ref disease and this clinical phenotype has previously been reported as 0 of 8 patients progression-free at 1 year following HDT-ASCT.8 With 47% of the subjects on our study having received 3 or more lines of prior therapy, secondary analysis of a similar group on the CORAL study revealed <40% OS at 2 years in patients requiring a second salvage (3 lines of therapy) prior to HDT-ASCT.27 The recently reported Scholar-1 study demonstrated <20% OS at 2 years in patients who were refractory to first and subsequent salvage therapy.28 Two other groups, City of Hope (CoH) and MD Anderson Cancer Center (MDACC), have investigated CAR T cells directed against CD19 (CAR19) post–HCT-ASCT in consolidation for B-NHL. The CoH group reported 2 sequential phase 1 studies and 16 patients treated in total.29 In their initial study, they observed a 1-year PFS of 50% (n = 8) using central memory T cells transduced with a CAR lacking a costimulatory domain. Upon incorporating a CD28 costimulatory domain into the central memory T cell CAR19 construct (n = 8), they observed a 1-year PFS of 75% in their subsequent study. Of note, in contrast to our study, 9 of 16 of their patients were in CR at time of HDT-ASCT and their second study included 4 of 8 patients with mantle cell lymphoma in first CR with expected median PFS exceeding 10 years,30 likely explaining the disparity in outcomes of these small patient cohorts. The group from MDACC enrolled 9 patients and treated 7 of them following HDT-ASCT using a transposase-based delivery system (SleepingBeauty) to generate CD28 costimulated CAR19 T cells. Six of the 7 subjects, including various histologies and 3 FDG-PET− subjects at time of HDT-ASCT, remained in remission at a median follow-up exceeding 2 years.31
There are many potential mechanisms of resistance to 19-28z CAR T-cell therapy in the 9 subjects who progressed on our study. In comparison with the NCI group that treated DLBCL patients with the same CAR-signaling module, no subjects on our study demonstrated persistence of the CAR T cells beyond 22 days whereas the NIH and ZUMA-1 studies detected peripheral blood 19-28z CAR T lasting months following infusion.10,12,14 The NCI group32 and others33 have previously reported the requirement for CD19 antigen burden to establish persistence and efficacy of CD19 CAR T cells. In our study, the limited persistence of 19-28z CAR T cells may result from subjects being in partial remission with depleted tumor and low endogenous CD19 antigen burden following salvage and myeloablative chemotherapy. These results question the rationale of using this therapy to consolidate remissions as opposed to treating overt rel/ref disease. Potential vaccinial strategies might serve to effectively circumnavigate the low endogenous CD19 antigen burden state of a consolidative platform and thus warrants investigation.34 Also, phosphoproteomic-analyzed differences between CD28 vs 4-1BB costimulatory CD8+ human CAR T cells have demonstrated faster changes in protein phosphorylation correlating with effector-like phenotype in CD28 constructs vs more memory-associated expression of genes in the 4-1BB product, which may contribute to greater persistence of 4-1BB CAR T.35 Additional strategies to enhance persistence include the use of “armored” CARs expressing additional signaling molecules and/or genes promoting autonomous secretion of lymphoproliferative cytokines with the intention of enhancing persistence and effector function.36,37 Lastly, CD19 antigen escape has been described with CD19 CAR therapy38 and 2 of the subjects who progressed on our study were found to be CD19− on the surface membrane at rebiopsy. CAR-modified T cells recognizing multiple antigens may serve as an effective strategy to counter this mechanism of resistance.39
We attempted to correlate serum cytokines and 19-28z CAR-modified peak expansion and cell persistence to toxicity and efficacy. The predominant toxicity was grade 3-4 neurotoxicity or seizure of any grade in 10 of the 15 subjects. Additionally, 6 of 10 of these subjects also experienced CRS. In the 10 subjects who experienced neurotoxicity, there was increased upregulation of IFN-γ and a trend toward increased upregulation of IL-10. Both IFN-γ9 and IL-109,10 have been correlated to severe neurotoxicity in studies of CAR19 in overt rel/ref DLBCL/B-NHL. Interestingly, no significant neurotoxicity or CRS was reported in the other 2 studies administering CAR19 following HDT-ASCT.29,31 With a very similar 19-28z CAR T construct, the NIH hav noted a decreased toxicity signal with reduced-intensity lymphodepletion10 in contrast to their earlier investigations of higher-dose conditioning.40,41 Parallel phase 2, multicenter studies resulting in US Food and Drug Administration (FDA)-approval demonstrate likely increased neurotoxicity with CD28 costimulation (axicabtagene ciloleucel)14 in contrast to 4-1BB (tisagenlecleucel).42 This difference might be attributable to more rapid, and intensive, signaling toward an effector T-cell–like transcriptional profile relative to 4-1BB costimulation,35 although the clinical phenotypic neurotoxicity disparity remains unclear. The neurotoxicity on our study may have also been contributed by the ubiquitous use of pegylated G-CSF on day +1 following HDT-ASCT, and prior to 19-28z CAR T infusion. Our group has previously described monocyte/macrophage-derived cytokine enrichment in blood and cerebrospinal fluid, including G-CSF and granulocyte macrophage colony-stimulating factor, with neurologic toxicity in acute lymphoblastic leukemia patients treated with 19-28z CAR-T.43 A recent report from the Mayo Clinic demonstrated that, in a xenograft model, neutralization with an anti–granulocyte macrophage colony-stimulating factor can abrogate neuroinflammation.44
We sought to immunophenotype the end of production 19-28z CAR T cells to establish correlations with outcomes. Somewhat surprisingly, higher percentages of naive-like CD45RA+CCR7+ T cells within both CD4 and CD8 subsets of the CAR T products correlated with greater likelihood of progression events. The NIH group, as well as others,45 has reported the merits of generating an optimized adoptive cellular product with naive-like CD45RA+ T cells, including T-cell central memory stem cells (CD45RA+CD62L+CCR7+CD95+) that display enhanced proliferative and survival capacity translating to improved efficacy compared with more differentiated, and shorter-lived, phenotypes.46,47 These data prompted a recent feasibility clinical trial exploring selection of less-differentiated immunophenotype T cells for CAR19 production and treatment in rel/ref pediatric acute lymphocytic leukemia with encouraging results.33 In our study, given the lack of CAR T-cell persistence potentially secondary to depletion of CD19 antigen in subjects with partial remission proceeding to HDT, it is possible that a 19-28z CAR T product composed of a higher proportion of differentiated, effector immunophenotype provides a relatively greater immediate benefit toward disease control as opposed to patients with larger-volume, active, refractory disease at the time of CAR19 wherein response may be more dependent on persistence of a naive-like phenotype CAR T product.48 This may serve as rationale toward reduced dosing, multiple infusions over multiple weeks as our group has used for allogeneic antiviral cytotoxic T lymphocyte therapy for Epstein-Barr virus–associated lymphomas with efficacy.49
In summary, we have demonstrated the feasibility of 19-28z CAR T administration following HDT-ASCT for rel/ref aggressive B-NHL. The therapy was met with a large burden of reversible neurotoxicity, potentially related to high-dose conditioning prior to 19-28z CAR T. The contribution of pegylated G-CSF is also potentially suspect. Although the long-term durability of this therapy was seen in only a few subjects, the high-risk characteristics of this relatively small phase 1 study population limits efficacy determination.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Memorial Sloan Kettering Society, Cycle for Survival, the American Society of Blood and Marrow Transplantation New Investigator Award, Juno Therapeutics, and National Institutes of Health, National Cancer Institute grant P30 CA008748. This work was also supported by the National Gene Vector Biorepository.
Authorship
Contribution: C.S.S., I.R., C.H.M., S.G., and R.J.B. designed, conducted, and analyzed the study and wrote the paper; B.S., A.N., T.P., M.H., A.N.S., Y.B., X.W., S.Y., A.D., Y.W., M.J.M., K.J.C., M.-A.P., J.P., and M.S. analyzed the data and wrote the paper; and C.S.S. and V.Z.S. created the visual abstract.
Conflict-of-interest disclosure: C.S.S., K.J.C., and J.P. have received consulting fees and funding support from Juno Therapeutics, Inc. I.R., M.S., and R.J.B. have received consulting fees and funding support from, and are inventors of patents licensed to, Juno Therapeutics, Inc, in which they have an equity interest. The remaining authors declare no competing financial interests.
Correspondence: Craig S. Sauter, Memorial Sloan Kettering Cancer Center, Box 276, 1275 York Ave, New York, NY 10065; e-mail: sauterc@mskcc.org.