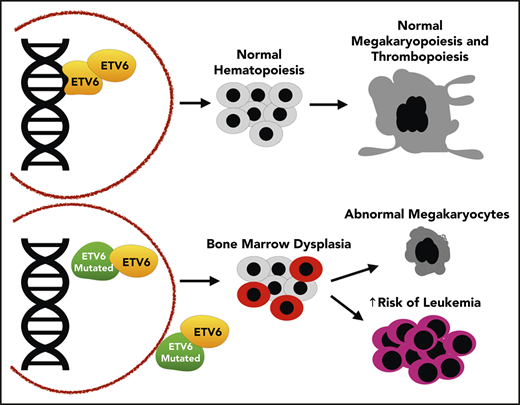
Abstract
Germ line mutations in ETV6 are responsible for a familial thrombocytopenia and leukemia predisposition syndrome. Thrombocytopenia is almost completely penetrant and is usually mild. Leukemia is reported in ∼30% of carriers and is most often B-cell acute lymphoblastic leukemia. The mechanisms by which ETV6 dysfunction promotes thrombocytopenia and leukemia remain unclear. Care for individuals with ETV6-related thrombocytopenia and leukemia predisposition includes genetic counseling, treatment or prevention of excessive bleeding and surveillance for the development of hematologic malignancy.
Introduction
It has become clear that a significant fraction of hematologic malignancies (HMs) arise in individuals with germ line alterations in cancer predisposition genes. This is highlighted by the inclusion of myeloid neoplasm with germ line predisposition in the 2016 World Health Organization’s classification of myeloid leukemia.1 Predisposition to HMs can be associated with increased risk of leukemia only or of solid tumors and leukemia or with systemic syndromes.2 A fourth group at increased risk of leukemia includes those with mutations in genes that also lead to thrombocytopenia and/or platelet function abnormalities. The purpose of this Blood Spotlight is to concisely review ETV6-related thrombocytopenia and leukemia predisposition disorder, which arises as a result of germ line mutations in ETV6.
Clinical features
ETV6-related thrombocytopenia and leukemia predisposition was first recognized in pedigrees with an autosomal dominant disorder of thrombocytopenia.3,4 Subsequent reports confirmed near-complete penetrance of thrombocytopenia,5-7 although some carriers with normal platelet counts have been reported.8-11 The thrombocytopenia is usually mild to moderate, with most patients having platelet counts >75 × 109/L. Complete blood counts (CBCs) usually show normal white blood cell counts and hemoglobin concentrations, with variably high mean corpuscular volumes and, less consistently, low mean platelet volumes (Figure 1; supplemental Table 1, available on the Blood Web site).

Abnormal bone marrow histology and CBCs in patients with ETV6-related thrombocytopenia and leukemia predisposition. (A) Abnormal cellular features (yellow arrows) with hyperchromatic small megakaryocytes, disseminated toxic granulations, and dysplastic eosinophils (magnification ×100; Wright-Giemsa stain). (B) Platelet count, white blood cell (WBC) count, hemoglobin (Hgb), mean cell volume (MCV), and mean platelet volume (MPV) in patients (PT) with germ line mutations in ETV6, as compared with related individuals (Rel) without mutation (Student t test).3-6,9,10
Abnormal bone marrow histology and CBCs in patients with ETV6-related thrombocytopenia and leukemia predisposition. (A) Abnormal cellular features (yellow arrows) with hyperchromatic small megakaryocytes, disseminated toxic granulations, and dysplastic eosinophils (magnification ×100; Wright-Giemsa stain). (B) Platelet count, white blood cell (WBC) count, hemoglobin (Hgb), mean cell volume (MCV), and mean platelet volume (MPV) in patients (PT) with germ line mutations in ETV6, as compared with related individuals (Rel) without mutation (Student t test).3-6,9,10
In addition to thrombocytopenia, carriers of ETV6 mutations have variable bleeding tendencies, with some patients not experiencing any bleeding despite low platelet counts and other patients having more bleeding than expected for mildly decreased platelet counts. Some have been reported to have abnormal platelet aggregation despite no major differences in platelet membrane receptor distribution (supplemental Table 1). Platelets from patients with ETV6 mutations exhibit abnormal platelet spreading on fibrinogen surfaces and abnormal clot retraction, suggesting a defect in platelet outside-in signaling.5,6 Bone marrow aspirates and biopsies from those without leukemia reveal dyserythropoiesis, megakaryocyte hyperplasia, and hypolobulated small megakaryocytes (Figure 1).3-5
Among the 96 cases from 23 pedigrees that have been reported and confirmed to carry an ETV6 mutation, 29 (30.2%) were diagnosed with HMs (supplemental Table 1). B-cell acute lymphoblastic leukemia (B-ALL) is the most common malignancy, accounting for approximately two-thirds of cases. Furthermore, in a study of >4000 children with ALL, germ line ETV6 variants were identified in almost 1%.10 Individuals with germ line ETV6 variants were more likely to have hyperdiploid leukemia, similar to recent findings with germ line IKZF1 variants, and were significantly older at diagnosis than those without.10,12 Other reported HMs in mutation carriers include myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), mixed-phenotype acute leukemia, diffuse large B-cell lymphoma, polycythemia vera, and multiple myeloma. Cooccurring mutations include those in BCOR, RUNX1, KMT2A, PAX5, and EPOR, but the extent to which these specifically cooperate with ETV6 dysfunction remains to be demonstrated.3,4 There may also be an increased risk of solid tumors, because at least 7 carriers have had various benign and malignant solid tumors at ages <50 years, and a common polymorphism in ETV6 is associated with an increased risk of colorectal carcinoma.13 Other nonhematologic associations are generally not shared across pedigrees.3,7,10 Type of mutation does not seem to influence the penetrance of malignancy.
ETV6 structure and function
ETV6 is a member of the ETS family of transcription factors encoded by the ETS variant 6 gene (ETV6), located on chromosome 12p13.2. ETV6 functions primarily as a transcriptional repressor in complex with SIN3A, NCOR, and HDAC3.14-16 The 57-kDa ETV6 protein has highly conserved PNT and ETS DNA-binding domains (Figure 2). PNT is essential for homo- and heterooligomerization, whereas the ETS domain is essential for DNA binding at the core 5′GGA(A/T)3′ sequence recognized by other ETS family members.17 The 2 domains are joined by a central linker domain,14 known to be involved in nuclear export,18,19 binding of NCOR and HDAC3,16 and transcriptional repression.20
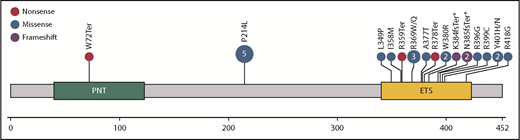
Schematic of ETV6 protein with functional domains and location of germ line mutations from 23 reported pedigrees. The number of families with alterations at the site is indicated in the circles if >1. A deletion that includes all of exon 2 has also been reported.44 *Germ line deletions in ETV6 involving splice sites.
Schematic of ETV6 protein with functional domains and location of germ line mutations from 23 reported pedigrees. The number of families with alterations at the site is indicated in the circles if >1. A deletion that includes all of exon 2 has also been reported.44 *Germ line deletions in ETV6 involving splice sites.
ETV6 is highly expressed in early hematopoietic progenitor cells (HPCs) and is essential for hematopoiesis in the bone marrow.21 Complete knockout of Etv6 was lethal to murine embryos by E11.5 because of a yolk sac angiogenesis defect,22 whereas targeted disruption revealed defects in hematopoiesis from bone marrow but not fetal liver.23,24 More specifically, conditional knockout of Etv6 impaired early HPC numbers and function but had little effect when deleted in committed lymphoid progenitors. In contrast, knockout of Etv6 in megakaryocyte progenitors led to dramatic decreases in platelet counts, suggesting a megakaryocyte differentiation and maturation defect.23 Although ETV6 has been characterized as a transcriptional repressor, only a few transcriptional targets of ETV6 have been reported,25-27 and interestingly, the megakaryocyte/platelet glycoprotein 1bα (GP1bα) and glycoprotein IX (GPIX) are among them.28,29 Additionally, ETV6 dimerizes with the ETS transcription factor FLI1, which is known to be involved in megakaryopoiesis,28 suggesting that abnormal interactions be-tween ETV6 and FLI1 contribute to thrombocytopenia.
ETV6 is involved in the most common somatic translocation in childhood B-ALL, resulting in the chimeric fusion protein ETV6-RUNX1, in up to 25% of cases.30 In these cases, the other ETV6 allele is often mutated or deleted subsequent to the translocation, implicating a tumor-suppressive function for ETV6, which is supported experimentally.27,31-34 Somatic point mutations or deletions are also recurrently found in high-risk B-ALL,35-38 immature T-cell ALL,39,40 and, less frequently, MDS/AML,41-43 further underscoring the importance of ETV6 in leukemogenesis.
Molecular and cellular consequences of ETV6 dysfunction
A majority of germ line mutations in ETV6 are missense variants in the DNA-binding domain of the protein (Figure 2), and most are private within pedigrees. However, there are 5 pedigrees reported with a recurrent mutation in the central domain (P214L), and some amino acids have multiple variants reported (Y401H/N, R369Q/W). The R369Q and R399C variants have reduced DNA binding,3 and all tested variants have impaired transcriptional repression and/or dominant-negative activity when tested with wild-type (WT) ETV6.3-5,7 Deletions have also been described, 3 of which alter splice sites and are predicted to result in protein truncation (K384fsTer and 2 resulting in N385fsTer).6,7,11,44 All tested variants have reduced nuclear and increased cytoplasmic localizations of ETV6.3,4,7 This suggests that ETV6 variants may impair WT ETV6 function in part by oligomerizing with and sequestering WT ETV6 out of the nucleus, although altered subcellular localization has yet to be demonstrated with endogenously expressed ETV6. Reduced association with corepressors may also alter transcriptional function, as demonstrated for the A377T and Y401N variants.5
Global patterns of gene expression are affected by the expression of mutant ETV6.3,4 In particular, changes in expression levels of genes involved in platelet biogenesis were pronounced, including MYL9, VWF, and GP9. Importantly, platelets from individuals with ETV6 mutations have decreased expression of CDC42 and RHOA, critical regulators of cytoskeletal dynamics in platelets.5 Interestingly, the ETV6 p.P214L platelet transcriptome showed an increase in erythroid-specific transcripts and decrease in platelet-specific transcripts as compared with unaffected relatives. The downregulation of platelet-specific genes suggests a function of ETV6 beyond direct transcriptional repression, perhaps by performing a yet-to-be-identified role as a transcriptional activator, showing lineage specificity with a repressive role in erythropoiesis and more permissive role in megakaryopoiesis, or affecting other transcription factor complexes. In accordance with these findings, we have shown an abundance of immature hypolobulated megakaryocytes in the bone marrow of patients with the p.P214L mutation,4 and megakaryocytes expressing ETV6 variants are smaller and form fewer proplatelets, whether patient derived or exogenously expressed.4-6
Management of patients with germ line ETV6 mutations
Recommendations from experts45-47 and consensus guidelines48 for the care of children and adults with an inherited thrombocytopenia and leukemia predisposition syndrome have been proposed. The differential diagnosis for suspected inherited thrombocytopenia and leukemia predisposition includes germ line mutation in ETV6, as well as RUNX1 and ANKRD26.2 Once the diagnosis has been made, management includes identifying related carriers, treatment and/or prevention of excessive bleeding, consideration of surveillance for the development of HMs, particularly MDS, and treatment of HMs, possibly including hematopoietic stem cell transplantation (HSCT). When possible, referral to a center with experience in managing higher volumes of patients with leukemia predisposition syndrome is optimal.45,48 Genetic counseling plays a critical role in the management of individuals with germ line ETV6 variants.49 Of particular importance is the counseling of siblings who may carry the same mutation and who may be considered as donors for HSCT.
Management of bleeding risk in individuals with ETV6 germ line mutation begins with a thorough assessment of personal and family bleeding histories, platelet count, and, if available, platelet aggregation studies. Because bleeding is usually mild, observation may be sufficient, whereas local measures, antifibrinolytic agents, and desmopressin can be considered if bleeding is more significant. Acute bleeding may be managed with platelet transfusions, but such transfusions are reserved for episodes of moderate to severe bleeding to mitigate the risk of alloimmunization, particularly in women of childbearing age.47 All these therapies can be used to prevent bleeding during hemostatic challenges such as surgery.
Surveillance for HMs can be achieved through serial measurement of CBC with differential (CBCd) and bone marrow aspirate and biopsy (BMA/Bx), although the benefit of doing so has yet to be demonstrated. CBCd every 3 to 6 months and BMA/Bx yearly has been suggested, but the burden of such evaluations must also be considered, particularly for young children and for a syndrome in which acute leukemia is more common than MDS and less likely to be detected before clinical presentation.45,46,48 BMA/Bx should include morphology, cytogenetics, fluorescence in situ hybridization, and molecular studies, which are particularly useful when the marrow morphology is abnormal because of the underlying disorder rather than the evolution of MDS/AML. Additional BMA/Bx should be considered in individuals with changes in CBCd that persist for 2 to 4 weeks.
Whether individuals with ETV6 deficiency disorder have different outcomes associated with leukemia compared with those with sporadic disease, including survival as well as immediate and late adverse effects of therapy, remains to be determined. These individuals are at greatest risk for B-ALL, the prognosis of which is excellent in childhood, and children with germ line ETV6 mutation and ALL had outcomes similar to those of children with germ line WT ETV6.10 Conversely, the prognosis for children and adults with MDS/AML is guarded, and hypothetically, the risk of a distinct second primary malignancy remains in the absence of HSCT, as a result of the ongoing presence of ETV6 mutations in HPCs. Therefore, consultation with a specialist in HSCT early in the course of HMs should be considered. Potential related donors should be counseled and tested because of the risk of donor-derived leukemia.50,51
Future directions
As with all newly defined rare diseases, there is much work to be done to improve the understanding and management of ETV6-related thrombocytopenia and leukemia predisposition. Critical in this regard is increased awareness among clinicians about this and other inherited thrombocytopenia and leukemia predisposition syndromes, as well as access to high-quality diagnostic laboratories. There is increasing availability of multigene panel testing for cancer predispositions and platelet disorders, but not all include ETV6. Furthermore, optimal testing for germ line mutations predisposing to leukemia is obtained from cultured fibroblasts46 ; however, this type of testing is not offered by many laboratories. A major challenge for diagnostic laboratories and clinicians is the classification of variants, which is especially difficult for rare genetic diseases with evolving phenotypes. Inconsistencies between laboratories in classification of variant pathogenicity further cloud the use of the information in clinical practice. The development of guidelines for variant classification through a collaboration between ClinGen and the American Society of Hematology in genes associated with inherited thrombocytopenia and leukemia predisposition, including ETV6, is under way. Lastly, there are efforts in multiple laboratories worldwide to better understand the molecular mechanisms of abnormal platelet and leukocyte development as well as predisposition to malignancies resulting from ETV6 dysfunction.
The online version of this article contains a data supplement.
Acknowledgment
J.D.P. receives research funding from the National Heart, Lung and Blood Institute, National Institutes of Health (HL120728).
Authorship
Contribution: J.D.P. and C.C.P. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jorge Di Paola, 12800 East 19th Ave, Mail Stop 8302, Aurora, CO 80045; e-mail: jorge.dipaola@ucdenver.edu; and Christopher C. Porter, 1760 Haygood Dr, E370, Atlanta, GA 30030; e-mail: chris.porter@emory.edu.