

BACKGROUND:
T-cell prolymphocytic leukemia (T-PLL) is a rare, aggressive, post thymic lymphoid malignancy, with an incidence of approximately 0.1/100,000 people. T-PLL accounts for ~2% of all mature lymphocytic leukaemias in adults >30 years. Median overall survival (OS) ~20 months and long term remissions are infrequent. Intravenous alemtuzumab, a monoclonal antibody directed against CD52 remains the most effective treatment in T-PLL. At the Royal Marsden Hospital (RMH) we have been using alemtuzumab for T-PLL since the early 1990s. We describe our experience over the last 3 decades.
METHODS:
We included 174 T-PLL patients that were diagnosed or treated at RMH between 1989-2019. Immunophenotyping data was available through our hematological malignancy diagnostic service (HMDS) from 1998 onwards. Kaplan-Meier analysis was used for OS and disease free interval (DFI)
RESULTS:
There were 174 patients in total. Mean age at diagnosis was 61 years old (range 32-88) and M: F ratio was ~2:1 (113:61). 90 patients had reliable information on complete blood count (CBC) at diagnosis with median white blood cell count of 74 x 109/L (range 10-918), median hemoglobin 126 g/L (range 59-175) and median platelet count 116 x 109/L (range 7-513).
Sufficient immunophenotyping data was available for analysis in 135/174 patients. The results showed predictably high expression of CD2, CD3, CD5, CD7, CD52 and TCRαβ. CD25 was positive in 67/135 (50%) of cases which is higher than the 18-35% seen in the current literature. CD4+/CD8- cases comprised 83/135 (61%) with CD4-/CD8+ 19/135 (14%), CD4+/CD8+ 32/135 (23%) and CD4-/CD8- 2/135 (2%). These results are summarized in table 1.
Karyotyping/FISH (fluorescent in-situ hybridization) records showed a clonal result in 84 patients and of these 65/84 (77%) had an aberration of chromosome 14. These comprised 48/65 with inv(14), 6/65 t(14;14), 2/65 t(X;14), 1/65 TCL1 gene rearrangement by FISH and 8/65 with TRA/TRD involvement by FISH. Other frequent abnormalities seen were i(8)(q10) in 31/84 (39%) and complex karyotypic abnormality in 40/84 (48%). Lower frequency abnormalities seen were monosomy 11 in 8/84 (9%), monosomy 12 in 8/84 (9%), trisomy 8 in 6/84 (7%), 17p loss in 5/84 (6%) and mononsomy 13 in 4/84 (5%).
Alemtuzumab was used in 116 patients, 69/116 (59%) receiving it as frontline treatment and 47/116 (41%) as salvage therapy. In 50/116 (43%) alemtuzumab was used as single agent, the remaining 66/116 (57%) receiving combination therapy, mostly pentostatin.
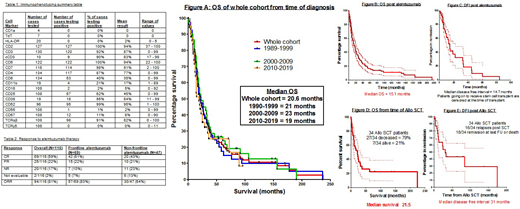
Mean time from diagnosis to receiving alemtuzumab was 6.8 months (range 0-53 months). Overall response rate (ORR) to alemtuzumab was 94/116 (81%) with complete remission (CR) 69/116 (59%), partial remission (PR) 25/116 (22%) and no response (NR) 20/116 (17%). Alemtuzumab produced better response rates when used as frontline therapy. Table 2 summarizes these results and OS and DFI are shown in figures B and C.
Allogeneic stem cell transplant (allo-SCT) was performed in 34 patients. Median OS post allo-SCT was 22 months and median DFI 31 months. Relapse rate post allo-SCT was 47%, non-relapse mortality 38% and transplant related mortality 29%. Figures D and E show OS and DFI post allo-SCT. Although relapse rates are high post allo-SCT there is a small cohort of patients who are achieving long term remission.
We analysed OS by decade of diagnosis covering 3 decades 1990-1999, 2000-2009 and 2010-2019. The median OS for the whole cohort was 20.6 months with median OS of 21 months, 23 months and 19 months for each decade respectively. Results are shown in figure A. There was no statistically significant difference between the curves by log-rank analysis.
DISCUSSION:
Immunophenotyping results were similar to previous published data on T-PLL except for an increase in CD25 expression at 50%. Our data shows that outcomes in T-PLL have remained unchanged since the first decade of alemtuzumab usage, with no improvement in OS in the last 20 years. Despite improvements in diagnostic techniques and supportive care median OS is static at approximately 20 months.
These results highlight the need for novel therapies in T-PLL. Given the rarity of T-PLL, international, multi-center, randomised trials are needed to improve outcomes.
Cross:Royal Marsden Cancer Charity: Other: MD Residency, Research Funding. Iyengar:Abbvie: Honoraria; Janssen: Honoraria. Dearden:Janssen: Honoraria; Genentech: Honoraria; Abbvie: Honoraria; Sanofi: Honoraria.

