

Asparaginase is an antileukemic enzyme that depletes the nonessential amino acid asparagine, but resistance is a common clinical problem whose biologic basis is poorly understood. We recently found that Wnt-induced inhibition of glycogen synthase kinase 3 (GSK3) profoundly sensitizes drug-resistant leukemias to asparaginase (Hinze et al, Cancer Cell, 2019;35:664). This effect is mediated by a β-catenin independent branch of Wnt signaling termed Wnt-dependent stabilization of proteins (Wnt/STOP), which inhibits GSK3-dependent protein ubiquitination and proteasomal degradation (Acebron et al, Mol Cell, 2014;54:663). Thus, asparaginase-resistant leukemias rely on catabolic protein degradation as an alternative source of amino acids to survive asparaginase therapy. Asparaginase resistance is selectively mediated by GSK3α, because its genetic or pharmacologic inhibition fully phenocopied Wnt-induced sensitization to asparaginase (p < 0.0001), whereas selective inhibition of GSK3β had no effect. This is surprising because GSK3α and GSK3β are closely related paralogs thought to be redundant for many of their biologic functions. Thus, our objective was to define why asparaginase resistance is selectively dependent on GSK3α activity.
To define the GSK3 domains responsible for asparaginase resistance, we leveraged the fact that selective depletion of GSK3α induces profound sensitization to asparaginase, and this effect is rescued by expression of a cDNA encoding GSK3α, but not GSK3β. We thus tested whether asparaginase resistance could be restored by expression of a series of GSK3 alleles in which the N-terminal, kinase, and C-terminal domains of GSK3α and GSK3β were swapped in various configurations. This revealed that asparaginase resistance is dependent on the N-terminal domain of GSK3α, whereas the kinase and the C-terminal domain were interchangeable. Fusing the N-terminus of GSK3α to the kinase and C-terminal domains of GSK3β fully restored asparaginase resistance in GSK3α depleted T-ALL (p < 0.0001) and AML cells (p < 0.0001). By contrast, fusing the N-terminus of GSK3β to the kinase and C-terminus of GSK3α had no discernible effect on response to asparaginase (p = n.s.). To investigate how the N-terminus of GSK3α regulates asparaginase response, we first applied structural prediction algorithms. This revealed that the N-terminal domain of GSK3α is a low-complexity (or prion-like) domain predicted to be intrinsically disordered, features associated with liquid-liquid phase separation. Phase separation is an increasingly recognized feature of cell biology that allows cells to concentrate components of important biochemical reactions in so-called membraneless organelles, thus promoting high-catalytic efficiency. Indeed, immunofluorescence confocal microscopy revealed that GSK3α, but not GSK3β, translocates into cytoplasmic bodies in response to asparagine depletion (p < 0.0001). The cytoplasmic GSK3a bodies were membraneless, as assessed by a proteinase K protection assay, and appeared to be distinct from known phase-separated compartments such as stress granules, P-bodies and aggresomes. However, cytoplasmic GSK3α bodies colocalized with the heat shock protein 70 (HSP70), K48-linked ubiquitin and the proteasome, suggesting that these bodies function in protein unfolding, ubiquitination and degradation. Indeed, genetic depletion of either GSK3α or HSP70 blocked formation of phase-separated GSK3α bodies (p < 0.0001) and induced asparaginase sensitivity (p < 0.0001).
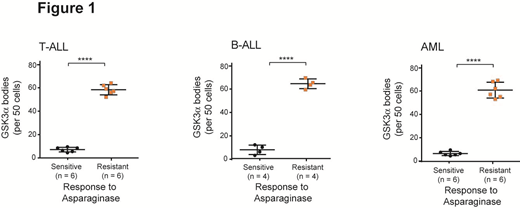
To explore the clinical relevance of GSK3α body formation in response to asparagine starvation, we tested a panel of matched asparaginase resistant vs. sensitive T-ALL, AML and B-ALL patient-derived xenografts (PDXs), and found that the ability of GSK3α to undergo phase separation significantly correlated with resistance to asparaginase in T-ALL, B-ALL and AML (Figure 1, p < 0.0001).
Our data support a model in which inducible phase separation of GSK3α and heat shock proteins represents a previously unrecognized response to amino acid starvation that concentrates the cellular machinery for protein degradation, thus allowing efficient catalysis of this alternative source of amino acids in response to amino acid starvation.
Chiosis:Samus Therapeutics: Equity Ownership, Patents & Royalties: Intellectual rights to the PU-FITC assay. Stevenson:Celgene: Research Funding. Neuberg:Celgene: Research Funding; Pharmacyclics: Research Funding; Madrigal Pharmaceuticals: Equity Ownership. Bourquin:Servier: Other: Travel support.

