

Acute myeloid leukemia (AML) is a myeloid progenitor-derived neoplasm of poor prognosis, particularly among the elderly, in whom age and comorbidities preclude the use of intensive therapies. Novel therapeutic approaches for AML are therefore critically needed. Adenosine monophosphate (AMP) activated protein kinase (AMPK) is a pleiotropic serine/threonine kinase promoting catabolism that represses anabolism and enhances autophagy in response to stress1. AMPK heterotrimers comprise catalytic α- and regulatory β- and γ-subunits, the latter harboring binding sites for AMP. Targets of AMPK include a host of metabolic pathway enzymes mediating carbohydrate, lipid and protein synthesis and metabolism. Accumulating evidence implicates AMPK in cancer biology, primarily as a tumor suppressor, although minimal AMPK activity may also be required for cancer cell growth under stress conditions2,3. Pharmacological activation of AMPK thus represents an attractive new strategy for targeting AML. We previously used the selective small molecule AMPK activator GSK621 to show that AMPK activation induces cytotoxicity in AML but not in normal hematopoietic cells, contingent on concomitant activation of the mammalian target of rapamycin complex 1 (mTORC1)4. However, the precise mechanisms of AMPK-induced AML cytotoxicity have remained unclear.
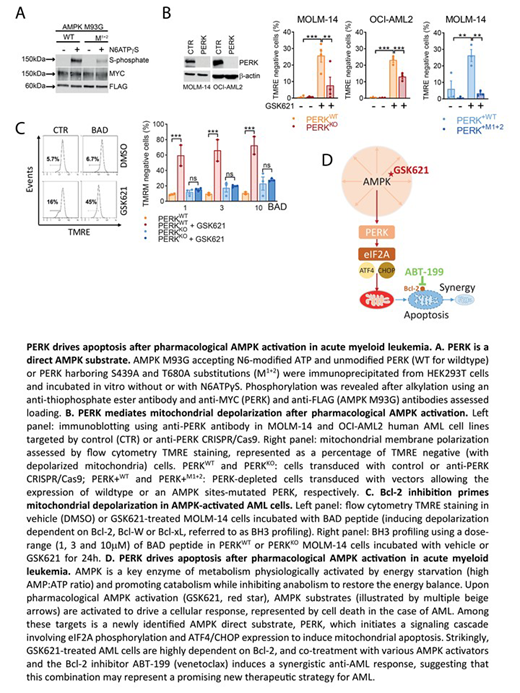
We integrated gene expression profiling and bioinformatics proteomic analysis to identify the serine/threonine kinase PERK - one of the key effectors of the endoplasmic reticulum stress response - as a potential novel target of AMPK. We showed that PERK was directly phosphorylated by AMPK on at least two conserved residues (serine 439 and threonine 680) and that AMPK activators elicited a PERK/eIF2A signaling cascade independent of the endoplasmic reticulum stress response in AML cells. CRISPR/Cas9 depletion and complementation assays illuminated a critical role for PERK in apoptotic cell death induced by pharmacological AMPK activation. Indeed, GSK621 induced mitochondrial membrane depolarization and apoptosis in AML cells, an effect that was mitigated when cells were depleted of PERK or expressed PERK with a loss of function AMPK phosphorylation site mutation. We identified the mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2) as a downstream target of the AMPK/PERK pathway, as its expression was enhanced in PERK knockdown AML cells. Moreover, selective pharmacologic activation of ALDH2 by the small molecule ALDA-1 recapitulated the protective effects of PERK depletion in the face of pharmacological AMPK activation. Corroborating the impact of the AMPK/PERK axis on mitochondrial apoptotic function, BH3 profiling showed marked Bcl-2 dependency in AML cells treated with GSK621. This dependency was abrogated in PERK-depleted cells, suggesting a role for PERK in mitochondrial priming to cell death. In vitro drug combination studies further demonstrated synergy between the clinical grade Bcl-2 inhibitor venetoclax (ABT-199) and each of four AMPK activators (GSK621, MK-8722, PF-06409577 and compound 991) in multiple AML cell lines. Finally, the addition of GSK621 to venetoclax enhanced anti-leukemic activity in primary AML patient samples ex vivo and in humanized mouse models in vivo. These findings together clarify the mechanisms of cytotoxicity induced by AMPK activation and suggest that combining pharmacologic AMPK activators with venetoclax may hold therapeutic promise in AML.
References
1. Lin S-C, Hardie DG. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metabolism. 2018;27(2):299-313.
2. Hardie DG. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clinical Cancer Research. 2015;21(17):3836-3840.
3. Jeon S-M, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch. Pharm. Res. 2015;38(3):346-357.
4. Sujobert P, Poulain L, Paubelle E, et al. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep. 2015;11(9):1446-1457.
Tamburini:Novartis pharmaceutical: Research Funding; Incyte: Research Funding.

