

Background: AML with myelodysplasia related changes (AML-MRC) is as specific WHO category with poor prognosis. It requires ≥ 20% of blasts, and (1) the history of MDS or MDS/MPN, or (2) "MDS related cytogenetic abnormalities", or (3) multilineage dysplasia. Drugs such as Vyxeos® have been approved by FDA and EMA only for treatment of t-AML or AML-MRC. However, counting blasts or grading dysplasia in clinical routine is hampered by limited reproducibility due to different levels of expertise and small phenotypic alterations, challenging upfront treatment decisions. Cytogenetics is not available in all cases and has 5-10 days of turnaround time (TAT). In contrast, next-generation sequencing (NGS) panels for AML are now broadly available at faster TAT.
Aim: (1) Use machine learning to define a molecular AML-MRC signature; (2) compare the impact of conventional WHO definitions and molecular factors on classification and outcome.
Patients and Methods: Gold standard routine AML diagnosis was performed on 739 cases. Overall survival (OS) data was available for 619 patients. Amplification-free whole genome sequencing was performed on HiSeqX and NovaSeq with median coverage of 106x. Gender-matched reference DNA was used for unmatched normal variant calling with Strelka2. Pindel was used for FLT3-ITD. For variant classification, we applied a GnomAD cutoff of 0.0005 and filtered on protein-truncating and (likely) pathogenic variants from databases.
Results: According to WHO standards 165/739 (22%) cases fulfilled MRC criteria (96 male; 69 female). The non-MRC cohort (n=574) represents a heterogeneous AML population incl. the WHO defined recurrent cytogenetic abnormalities or t-AML (301 male, 273 female). Median age was higher in the MRC cohort (73 [22-90] vs. 64 [18- 93] years, p<.001) and OS was significantly shorter (median 6 vs. 23 months, p<.001).
Mutation analysis was limited to 73 frequently mutated genes, in order to allow application of our model on prospective diagnostic cases analyzed by common routine panels. In the MRC group, up to seven mutations were found per patient and an average of 2.7 genes per patient were mutated. The most frequently mutated gene in AML-MRC was TP53 (62/165, 38%) as expected by the inclusion of complex karyotypes. TP53 mutations were associated with shorter OS in the MRC cohort (median: 3 vs. 11 months, p =.001).
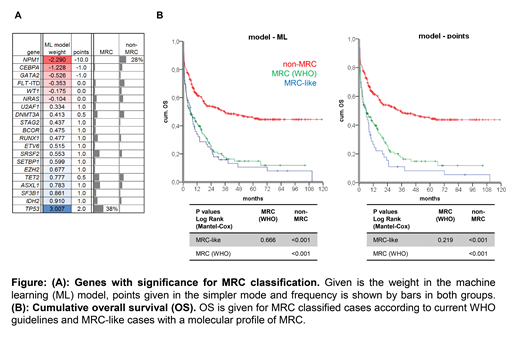
We used machine learning (ML) approaches to identify with LASSO regression and 10-fold cross-validation the most informative features to distinguish between MRC and patients without MRC. The dataset was randomly divided into a training (90%) and test set (10%) and the procedure was repeated 500 times to cover all the variance in the dataset and to extract the most reliable factors. Factors with the highest weight on AML-MRC prediction were mutations in TP53, RUNX1, SETBP1, splicing factors and epigenetic regulators, and absence of mutations in NPM1, CEBPA and others (s. figure). In order to allow our model to be used in a routine diagnostic workflow, we also used the genes identified by ML but classified mutations by a simpler point system (≥2 points as cutoff for MRC, s. figure). This allowed us to identify 83% (137/165 by ML) and 70% (116/165 by points) of cases currently defined as MRC solely by molecular genetics. Including cytogenetic data and patient's history in an informed genetic model results in 99% (164/165 by ML) and 96% (159/165 by points) of true positive MRC definition.
However, the molecular models classified 112 (ML) and 80 (points) of the 574 non-MRC cases, as being AML-MRC. Even after excluding AML with recurrent cytogenetic abnormalities and t-AML, 14% (82/574 DL) or 11% (63/574 points) show a MRC-like molecular profile. In both models MRC-like patients had dismal outcome analogous to AML-MRC (median OS: 6 months for both) and significantly inferior to remaining non-MRC patients (6 vs. 35 months, s. figure).
Conclusions: (1) Using patients' history and genetic information instead of morphology allow to identify 96-99% of AML-MRC as defined in WHO today. In the future, extended NGS panels (e.g. incl. fusion gene detection) will allow fast and standardized AML-MRC classification even without chromosome banding analysis. (2) The molecular MRC-like pattern can be found in >10% of patients currently not classified as AML-MRC but with comparably poor OS. This suggests considering MRC treatment strategies for patients with MRC-like molecular profile.
Baer:MLL Munich Leukemia Laboratory: Employment. Walter:MLL Munich Leukemia Laboratory: Employment. Stengel:MLL Munich Leukemia Laboratory: Employment. Hutter:MLL Munich Leukemia Laboratory: Employment. Meggendorfer:MLL Munich Leukemia Laboratory: Employment. Kern:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership.

