

Introduction: Due to the range of biological and molecular heterogeneity in diffuse large B-cell lymphoma (DLBCL), personalized risk stratification and treatment is a promising avenue to improving outcomes. Although most risk stratification depends primarily on clinical data (e.g. IPI), the addition of molecular, genomic or disease burden (e.g. quantitative PET imaging) features in DLBCL could help better stratify patients (pts) according to disease biology or burden. Such data are often hard to obtain in routine clinical settings; current methods remain limited by the need for tissue samples and low reproducibility in daily practice. A single method to assess such molecular markers from plasma samples could enable a standardized process. Here, we use a circulating tumor DNA (ctDNA)-based next-generation sequencing (NGS) method on pre-treatment plasma samples from first-line DLBCL pts to show prognostic correlations from molecular and disease burden assessments.
Methods: We performed targeted NGS on plasma samples from 310 previously untreated DLBCL pts enrolled in the GOYA study (NCT01287741) with a custom DLBCL-specific panel using a workflow optimized for ctDNA. Cell-free DNA (cfDNA) was isolated from plasma and an average of 30.7ng (range, 0.7-50ng) of cfDNA was used. Library preparation and NGS were performed with a modified AVENIO ctDNA workflow, with a custom panel of ~314kb designed to cover regions relevant for cell-of-origin (COO) and minimal residual disease in DLBCL. Single nucleotide variants (SNVs), insertions/deletions (indels), and fusions were determined, and criteria based on publicly available data removed non-tumor specific variants. For each sample, number of tumor genome copies per mL of plasma (MMPM), a measure of tumor burden, was calculated using the allele fractions of variant calls and cfDNA mass. Variant calls from 230 pts were used to build a machine learning model to determine COO, which was tested on the remaining 80 pts. Variant and COO calls were correlated with data from corresponding tissue samples, including mutations from FoundationOne® Heme and gene expression-based COO determination from Lymph2Cx (NanoString). Variant, COO, and disease burden assessments from ctDNA NGS were also compared with clinical variables, including IPI, baseline quantitative PET imaging, and progression-free survival (PFS) data.
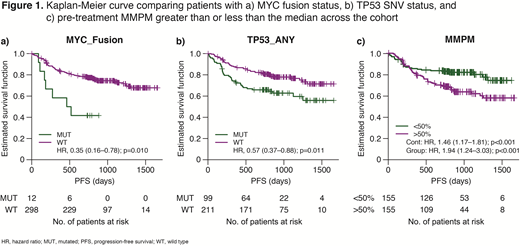
Results: 77% of SNVs (958/1247), 63% of indels (72/115), and 74% of fusions (49/66) detected with the FoundationOne assay were detected in the plasma samples. COO calls from plasma showed 93% concordance with Lymph2Cx calls from tissue when both methods classified a sample, with 37/40 germinal center B-cell (GCB) agreeing and 17/18 non-GCB agreeing in the test set. Subsequently, correlation of various metrics from the ctDNA assay with clinical outcomes was assessed. Non-GCB pts showed a trend towards worse PFS when compared with GCB pts (hazard ratio [HR], 1.23; 95th percentile: 0.79─1.92; p=0.32). Additionally, worse PFS was observed for pts with MYC fusions (n=12; HR, 2.83; 95th percentile: 1.29─6.21; p=0.010) and TP53 SNVs or indels (n=99; HR, 1.75; 95th percentile: 1.13─2.70; p=0.031), with a similar trend for BCL2 fusions (n=31; HR, 1.96; 95th percentile: 0.94─4.07; p=0.072). Baseline MMPM was significantly correlated with total metabolic tumor volume (TMTV) as measured by PET (r=0.36; p<0.001) and sum of the product of diameters (SPD; r=0.13; p=0.022). Higher MMPM values corresponded to higher IPI scores (ANOVA p<0.001), higher likelihood of bulky disease (p=0.019), and worse PFS as a continuous variable (HR, 1.46; p=0.0006; Figure 1). In a multivariate model accounting for TMTV, MMPM was not prognostic (p=0.15), but in a model containing SPD and MMPM, both were independently prognostic (MMPM p=0.010; SPD p=0.0046), suggesting that SPD and MMPM provide complementary prognostic information. Interestingly, in this sample set, activated B-cell samples also showed a significantly higher MMPM than GCB and unclassified samples (p=0.0017). Even when considering COO and IPI in a multivariate analysis, MMPM remained correlated with PFS (HR, 1.23; p=0.079).
Conclusions: We describe a single NGS-based method, which calls variants, determines COO, and assesses tumor burden from plasma. Using these results, we show that pre-treatment plasma-based molecular and tumor burden measurements in previously untreated DLBCL pts correlate with PFS.
Tabari:F. Hoffmann-La Roche: Equity Ownership; Roche Sequencing Solutions: Employment. Lovejoy:Roche Sequencing Solutions: Employment. Lin:Roche Sequencing Solutions: Employment; Veracyte: Other: Veracyte (spouse). Bolen:F. Hoffmann-La Roche: Equity Ownership; Genentech, Inc.: Employment. Saelee:Roche Sequencing Solutions: Employment. Lefkowitz:Roche Sequencing Solutions: Employment. Kurtz:Roche: Consultancy. Vitazka:Roche Sequencing Solutions: Employment. Venstrom:F. Hoffmann-La Roche Ltd: Employment. Nielsen:F. Hoffmann-La Roche Ltd: Employment, Equity Ownership. Parreira:F.Hoffmann-La Roche Ltd: Employment, Equity Ownership, Honoraria. Klass:Roche Sequencing Solutions: Employment; Roche: Equity Ownership. Luong:Roche Sequencing Solutions: Employment.

