

Introduction: Aplastic Anemia (AA) is diagnosis made after the exclusion of other acquired or inherited disease that can present with pancytopenia. In pediatric AA, there is often overlap with many other diseases including Paroxysmal Nocturnal Hemoglobinuria (PNH) and hypocellular myelodysplastic syndrome (MDS). The diagnosis is often difficult and can evolve. There are new tests and methodologies to guide diagnosis and treatment decision making.
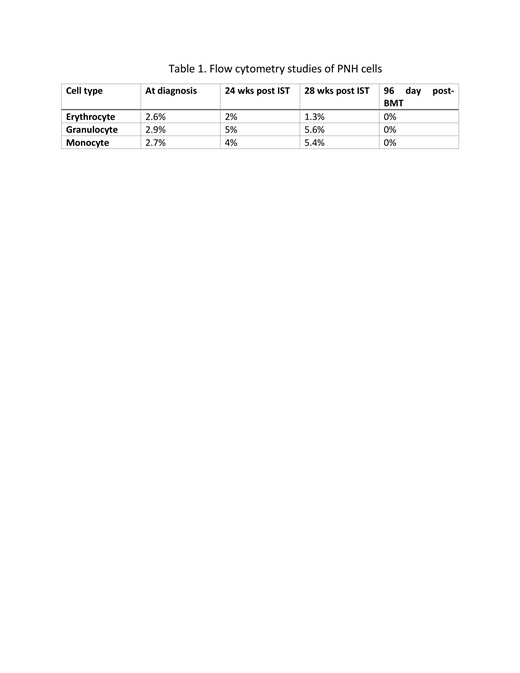
Case report: The patient is a previously healthy, 18-year-old Caucasian female referred for pancytopenia. Patient reported progressive fatigue and easy bruising for a month prior to evaluation. Initial white blood count was 2.4 x109/L, hemoglobin 9.7g/dl, platelets 39 x109/L, absolute neutrophil count (ANC) of 590, and absolute retic count 68 x 109/L. A bone marrow exam revealed a hypocellular marrow with 30% cellularity with no dysplasia or blasts on flow cytometry. Viral studies, folate and vitamin B12 levels were normal. Chromosomal analysis, Fluorescent In situ Hybridization (FISH) study for MDS, and telomere length assay were normal. A bone marrow failure (BMF) panel for sixty targeted genes showed no mutations associated with BMF. PNH studies by flow cytometry showed 2.6% erythroid, 2% granulocytes, 2.7% monocytes failed to express GPI associated anchoring proteins (GPI-APs) representing a small PNH clone, not unusual in patients with aplastic anemia (AA). Studies were c/w with moderate AA (mAA). Standard management of mAA is careful observation and treatment if the patient requires transfusions or ANC falls below 500. However, since our patient was soon starting college out of state, we decided to treat with Immunosuppressive Therapy (IST), consisting of equine Anti-thymocyte globulin (eATG) and Cyclosporine. At 24 weeks post IST, no response was observed and the patient was requiring more frequent platelet and red blood cell transfusions. Repeat PNH testing showed 2% erythroid, 5% monocytes and 4% granulocytes that were deficient in GPI-Aps (Table1). BM exam showed 60% cellularity that was consistent with a recovering marrow but there was relative erythroid hyperplasia, without evidence of dysplasia or blasts. At this time, with increasing marrow cellularity but persistent pancytopenia we began to strongly suspect MDS. However, repeat chromosomal analysis and FISH studies were normal and a Foundation One myeloid panel was negative for mutations associated with MDS. At 28 weeks post-IST, repeat BM exam revealed 70% cellularity, again with relative erythroid hyperplasia. At the same time PNH testing, showed that 1.3% erythrocytes, 5.6% granulocytes and 5.4% monocytes were negative for GPI-APs. We concluded that our patient evolved had from mAA to subclinical PNH/AA which accounted for the normocellular marrow. High Disease Activity (HDA) is defined as lactate dehydrogenase (LDH) ≥ 1.5 times upper limit of normal and is associated with large PNH clones. HDA can guide the use of eculizumab as pre-transplant therapy. (DeZern et al, 2012) Throughout the course of the disease, her LDH was normal. She did not have HDA and therefore, was not treated with eculizumab. Because of the normocellular marrow she was not eligible for CTN 1502-CHAMP study. However, the patient underwent reduced intensity conditioning (RIC) and a haplo paternal BMT, followed by post-transplant cyclophosphamide, as per CTN 1502- CHAMP. She tolerated her BMT well without major complications. She is now five months post-transplant, alive and well, fully engrafted with 100% donor chimerism and no signs of GVHD. Post-transplant PNH testing revealed all PNH clones were eliminated.
Conclusion: This case illustrates the diagnostic challenges of patients presenting with pancytopenia and the overlap and possible evolution of disease classifications; new diagnostic testing including gene mutation screens to aid in diagnosis; new concepts such as HDA in PNH to help guide treatment decision making; and the successful use of RIC and haplo BMT in a patient with PNH/AA and a normocellular BM
No relevant conflicts of interest to declare.

