Key Points
Neutrophil NADPH oxidase limits LTB4 production by controlling calcium influx.
LTB4 is a major driver in promoting neutrophilic inflammation in CGD mice in response to fungal cell walls.

Abstract
Leukocyte reduced NADP (NADPH) oxidase plays a key role in host defense and immune regulation. Genetic defects in NADPH oxidase result in chronic granulomatous disease (CGD), characterized by recurrent bacterial and fungal infections and aberrant inflammation. Key drivers of hyperinflammation induced by fungal cell walls in CGD are still incompletely defined. In this study, we found that CGD (CYBB−) neutrophils produced higher amounts of leukotriene B4 (LTB4) in vitro after activation with zymosan or immune complexes, compared with wild-type (WT) neutrophils. This finding correlated with increased calcium influx in CGD neutrophils, which was restrained in WT neutrophils by the electrogenic activity of NADPH oxidase. Increased LTB4 generation by CGD neutrophils was also augmented by paracrine cross talk with the LTB4 receptor BLT1. CGD neutrophils formed more numerous and larger clusters in the presence of zymosan in vitro compared with WT cells, and the effect was also LTB4- and BLT1-dependent. In zymosan-induced lung inflammation, focal neutrophil infiltrates were increased in CGD compared with WT mice and associated with higher LTB4 levels. Inhibiting LTB4 synthesis or antagonizing the BLT1 receptor after zymosan challenge reduced lung neutrophil recruitment in CGD to WT levels. Thus, LTB4 was the major driver of excessive neutrophilic lung inflammation in CGD mice in the early response to fungal cell walls, likely by a dysregulated feed-forward loop involving amplified neutrophil production of LTB4. This study identifies neutrophil LTB4 generation as a target of NADPH oxidase regulation, which could potentially be exploited therapeutically to reduce excessive inflammation in CGD.
Introduction
Leukocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a multisubunit enzyme assembled on plasma or phagosome membranes to generate superoxide, the precursor to reactive oxygen species (ROS) that is essential in pathogen killing and immunoregulation. These dual roles are reflected in clinical manifestations of chronic granulomatous disease (CGD), a primary immunodeficiency resulting from inactivating mutations in the NADPH oxidase and characterized by bacterial and fungal infections and aberrant inflammation.1 Even sterile fungal cell walls elicit increased neutrophilic inflammation associated with elevated proinflammatory cytokines in CGD mice,2-5 demonstrating that NADPH oxidase–derived ROS limit inflammation independent of antimicrobial activity. NADPH oxidase ROS impact multiple pathways triggered in response to fungal cell walls and other inflammatory stimuli, including attenuating activation of NF-κB and of the IL-1β inflammasome.1,3,6-9 However, key drivers of neutrophilic hyperinflammation in CGD remain incompletely defined.
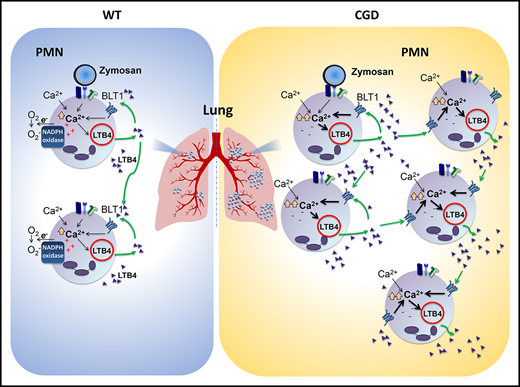
The inflammatory lipid mediator leukotriene B4 (LTB4) is an important initiator of neutrophilic inflammation.10-17 LTB4 is a potent neutrophil chemoattractant, enhances neutrophil-endothelial interactions, and activates neutrophil degranulation and ROS production.10 Neutrophils are a prominent source of LTB4 and also sense LTB4 through the high-affinity receptor BLT1, resulting in increased neutrophil LTB4 synthesis by both autocrine and paracrine routes.13,18,19 Neutrophil-derived LTB4 thus leads to the feeding forward of amplification of local LTB4 production, neutrophil recruitment, and swarming13-17 and can act as a signal-relay molecule from cell to cell.19,20 LTB4 is synthesized from arachidonic acid by the sequential action of cytosolic phospholipase A2α, 5-lipoxygenase (5-LOX), and leukotriene A4 hydrolase.21,22 Calcium plays a central role in LTB4 biosynthesis, and receptor-mediated neutrophil activation leads to a rapid increase in intracellular calcium, primarily by calcium influx from the extracellular milieu via store-operated calcium entry (SOCE).23 In turn, elevated intracellular calcium is needed to activate cytosolic phospholipase A2α and 5-LOX.21,22,24 Of note, NADPH oxidase–mediated electron transfer across the membrane leads to plasma membrane depolarization, which decreases the driving force for extracellular calcium entry.25-28 In the absence of the oxidase, the negative cell membrane potential in CGD neutrophils thus results in increased calcium influx and a relative overload of intracellular calcium,25-28 which is speculated to increase the proinflammatory activity of CGD neutrophils.28,29 Studies of how elevated calcium alters specific neutrophil responses, including its impact on LTB4 synthesis and in vivo relevance, are few.
Because LTB4 is an important early mediator of neutrophilic inflammation, we investigated whether NADPH oxidase deficiency affects LTB4 produced by neutrophils in vitro and whether LTB4 plays a role in the marked neutrophilic response induced by fungal cell walls in CGD. Mouse and human neutrophil generation of LTB4 in response to the sterile fungal cell wall particle zymosan or in response to immune complexes was amplified in the absence of the NADPH oxidase. This process correlates with increased intracellular calcium levels in CGD neutrophils and also necessitates cross talk between LTB4 and the BLT1 receptor. Moreover, in CGD mice, LTB4 played a major role in driving excessive neutrophil recruitment to the lung after pulmonary instillation of zymosan. This finding implicates dysregulated LTB4 production as promoting acute neutrophilic lung inflammation in response to fungal cell walls in CGD by an amplified feed-forward loop involving increased neutrophil production of LTB4.
Methods
Mice
Wild-type (WT) C57BL/6J and B6.SJL-PtrcaPep3b/BoyJ mice were purchased from The Jackson Laboratory or bred in house. Mice with inactivation of X-linked Cybb (X-linked CGD mice)30 on C57BL/6J or B6.SJL-PtrcaPep3b/BoyJ31 backgrounds were obtained from in-house colonies. Mice were maintained in specific pathogen-free conditions, and both males and females were used in experiments at between 10 to 14 weeks of age. All experiments were approved by the Institutional Animal Care and Use Committee at Washington University in St. Louis.
Mouse neutrophil isolation
Bone marrow (BM) was collected, and red blood cells were removed by hypotonic lysis and incubated in α-minimum essential medium (Thermo Fisher Scientific, Carlsbad, CA) with 2% heat-activated fetal bovine serum (MilliporeSigma, Burlington, MA) in a 10-cm tissue culture dish for 1 hour at 37°C in 5% CO2. Nonadherent cells were collected, and polymorphonuclear neutrophils (PMNs) were isolated with an EasySep Mouse Neutrophil Enrichment Kit (STEMCELL Technologies, Vancouver, BC, Canada). The purity of the PMNs was ≥95%.
LTB4 and calcium studies in murine neutrophils
Next, 96-well tissue culture plates were coated with 10% heat-inactivated fetal bovine serum. Neutrophils suspended in RPMI 1640 (0.4 mM Ca2+; Corning Inc., Corning, NY) without serum were added to the wells, with or without zymosan (ratio, 2 zymosan/1 PMN; MilliporeSigma) or immobilized immune complexes, and incubated at 37°C in 5% CO2 for various times. In experiments where extracellular calcium concentration was varied, PMNs were incubated in Hanks’ balanced salt solution in ambient air at 37°C. After centrifugation, supernatant was collected and stored in −80°C before LTB4 enzyme-linked immunosorbent assay (ELISA). Diphenyleneiodonium chloride (10 µM; DPI; InvivoGen, San Diego, CA), 10 µM U75302 (Cayman Chemical, Ann Arbor, MI), superoxide dismutase (SOD; MilliporeSigma), and/or catalase (MilliporeSigma) were added in some experiments. For calcium studies, BM PMNs were isolated by using a Percoll gradient, and intracellular calcium measurements were performed using flow cytometry.32
Neutrophil imaging in vitro
Mouse neutrophils were primed with mouse tumor necrosis factor-α, stained with Hoechst 33342, and incubated with fluorescein isothiocyanate-zymosan (Thermo Fisher Scientific) in a four-well microinsert dish for 1 hour before imaging with a Leica SP8X laser scanning confocal microscope, with a 10× Apochromat air objective (N.A. 0.4) in an Okolab Cage incubator.
Zymosan induced lung inflammation
Mice were challenged with 20 µg sterile zymosan by intranasal (IN) instillation and euthanized at various time points for analysis of inflammation. In some experiments, the zileuton or vehicle (100 mg/kg) was administered33 by gavage at various times, or 5 µg U7530234 or vehicle were given together with IN zymosan challenge, or the mice underwent neutrophil depletion as previously described.35
Statistics
For statistical analyses, we used Prism 8.0 (GraphPad Software Inc, San Diego, CA). Statistical significance was assessed by paired t test, Student t test, ratio paired t test, or 1-way ANOVA as indicated. A value of P < .05 was considered significant. Additional methods and details can be found in the supplemental Methods.
Results
PMNs have increased LTB4 production in vitro in the absence of NADPH oxidase activity, which correlated with increased intracellular calcium levels
We compared LTB4 production by WT and CGD neutrophils stimulated with zymosan, a particulate preparation of yeast cell walls, which activates neutrophils through dectin-1 and CD11b/CD18.36 CGD neutrophils generated ≈10-fold more LTB4 (Figure 1A) by 30 minutes of incubation, with levels plateauing after 1 hour. These results were not accounted for by differences in the fraction of zymosan-associated neutrophils between genotypes, and ≈10% of neutrophils had either bound or ingested zymosan by 30 minutes (supplemental Figure 1A-B, available on the Blood Web site). In the presence of the NADPH oxidase inhibitor DPI, zymosan-activated WT murine neutrophils also yielded higher amounts of LTB4, as did DPI-treated human neutrophils from healthy donors (supplemental Figure 1C-D). Neutrophils from a CGD patient, with a null CYBB mutation also generated more LTB4 after either zymosan or serum-opsonized zymosan stimulation compared with healthy donors (supplemental Figure 1D-E). The addition of the oxidant scavengers SOD and catalase resulted in only a modest increase in LTB4 for zymosan-activated WT mouse neutrophils (Figure 1B). Thus, degradation by NADPH oxidase-derived ROS by WT neutrophils only partially accounted for the higher LTB4 levels measured in CGD neutrophils. Reactive chlorine species also did not appear to play a role. Inhibiting myeloperoxidase (MPO) with 4-aminobenzoic acid hydrazide did not affect the LTB4 levels (supplemental Figure 1F). LTB4 levels from zymosan-stimulated neutrophils were unchanged by addition of exogenous H2O2 or sustained H2O2 generation by glucose/glucose oxidase, with or without MPO (supplemental Figure 1G). Incubation with H2O2 or H2O2/MPO also did not reduce the LTB4 present in supernatants from zymosan-stimulated neutrophils (supplemental Figure 1H).
![CGD mouse neutrophils had increased LTB4 production and elevated calcium influx compared with WT, and oxidant scavengers did not increase LTB4 in WT neutrophils. (A) Mouse BM neutrophils (4 × 106/mL) were stimulated for various times with zymosan (MOI = 2 zymosan: 1 PMN) in RPMI 1640 (0.4 mM Ca2+; n ≥ 3). *P < .05; ****P < .0001), by Student t test. (B) Mouse BM neutrophils (4 × 106/mL) were pretreated with 200 U/mL SOD and 200 U/mL catalase for 10 minutes and then stimulated for 1 hour with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 200 U/mL SOD and 200 U/mL catalase. n = 4 per group from 2 separate experiments. *P < .05, by paired t test. NS, not significant. (C-D) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils (1 × 106/mL) loaded with 3 µM indo-1 were stimulated with (C) Zymosan (100 µg/mL; MOI ≈ 10) with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Mean ± standard error of the mean from 2 to 4 independent experiments. *P < .05; **P < .01, by Student t test. (E) Mouse BM neutrophils (4 × 106/mL) were stimulated for 1 hour with zymosan (MOI = 2) in Hanks’ balanced salt solution with the indicated amounts of extracellular calcium. LTB4 levels in culture supernatant was measured by ELISA. n ≥ 3 per group from 3 separate experiments. Data are means ± SEM. *P < .01; ***P < .001, by Student t test. MOI, multiplicity of infection.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/12/10.1182_blood.2019003525/4/m_bloodbld2019003525f1.png?Expires=1769089294&Signature=SY3tE2DqlIBuf2SrKZG6J5cMYftt1eSs9lK-KJetuWLM1louaklb0oOx51xe9bgcfiivAsPDLlPWmAsecbMejnIBgRqMWG3L1lvospU7HsNjwc71Lzju0cAkz-c8w3f-yemMaHKoHM9FcrpcpWKqKEtTQw5yVrgHyC-mGZW58nwmltgpHNOYCfzTmCTyHByexI9C78EPtSUDTwsV66iNLKJF9KIc1X5-DDQOD8c1gXMGpfhCxVYMbyVky3tWlfm34s~ksF902JlJQ~AEEAVvbmtC7r6RZTRci8DQ~YkZUgUFKqHKgFNcGtNoZne2mei06BrAW5WPuj1mf-58a1VDdw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CGD mouse neutrophils had increased LTB4 production and elevated calcium influx compared with WT, and oxidant scavengers did not increase LTB4 in WT neutrophils. (A) Mouse BM neutrophils (4 × 106/mL) were stimulated for various times with zymosan (MOI = 2 zymosan: 1 PMN) in RPMI 1640 (0.4 mM Ca2+; n ≥ 3). *P < .05; ****P < .0001), by Student t test. (B) Mouse BM neutrophils (4 × 106/mL) were pretreated with 200 U/mL SOD and 200 U/mL catalase for 10 minutes and then stimulated for 1 hour with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 200 U/mL SOD and 200 U/mL catalase. n = 4 per group from 2 separate experiments. *P < .05, by paired t test. NS, not significant. (C-D) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils (1 × 106/mL) loaded with 3 µM indo-1 were stimulated with (C) Zymosan (100 µg/mL; MOI ≈ 10) with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Mean ± standard error of the mean from 2 to 4 independent experiments. *P < .05; **P < .01, by Student t test. (E) Mouse BM neutrophils (4 × 106/mL) were stimulated for 1 hour with zymosan (MOI = 2) in Hanks’ balanced salt solution with the indicated amounts of extracellular calcium. LTB4 levels in culture supernatant was measured by ELISA. n ≥ 3 per group from 3 separate experiments. Data are means ± SEM. *P < .01; ***P < .001, by Student t test. MOI, multiplicity of infection.
CGD mouse neutrophils had increased LTB4 production and elevated calcium influx compared with WT, and oxidant scavengers did not increase LTB4 in WT neutrophils. (A) Mouse BM neutrophils (4 × 106/mL) were stimulated for various times with zymosan (MOI = 2 zymosan: 1 PMN) in RPMI 1640 (0.4 mM Ca2+; n ≥ 3). *P < .05; ****P < .0001), by Student t test. (B) Mouse BM neutrophils (4 × 106/mL) were pretreated with 200 U/mL SOD and 200 U/mL catalase for 10 minutes and then stimulated for 1 hour with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 200 U/mL SOD and 200 U/mL catalase. n = 4 per group from 2 separate experiments. *P < .05, by paired t test. NS, not significant. (C-D) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils (1 × 106/mL) loaded with 3 µM indo-1 were stimulated with (C) Zymosan (100 µg/mL; MOI ≈ 10) with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Mean ± standard error of the mean from 2 to 4 independent experiments. *P < .05; **P < .01, by Student t test. (E) Mouse BM neutrophils (4 × 106/mL) were stimulated for 1 hour with zymosan (MOI = 2) in Hanks’ balanced salt solution with the indicated amounts of extracellular calcium. LTB4 levels in culture supernatant was measured by ELISA. n ≥ 3 per group from 3 separate experiments. Data are means ± SEM. *P < .01; ***P < .001, by Student t test. MOI, multiplicity of infection.
As LTB4 biosynthesis is strongly regulated by calcium, we examined zymosan-activated calcium influx. Intracellular calcium rose rapidly in the CGD neutrophils, compared with levels in the WT cells, and reached significantly higher levels across a range of extracellular calcium concentrations (Figure 1C-D). In contrast, there was no difference in calcium influx between WT and CGD neutrophils with agents that induce calcium influx but did not activate the NADPH oxidase, including pharmacological store depletion with thapsigargin or ionomycin (supplemental Figure 1I). However, activating the NADPH oxidase with phorbol myristate acetate in ionomycin-treated WT cells suppressed calcium influx compared with CGD neutrophils (supplemental Figure 1I). Thus, similar to human neutrophils,25,26 where the electrogenic effects of NADPH oxidase activity limit calcium influx, activated CGD mouse neutrophils displayed increased intracellular calcium levels.
Extracellular calcium levels similarly influenced zymosan-induced LTB4 production. LTB4 was not produced in the absence of extracellular calcium (Figure 1E). WT neutrophils generated increasing amounts of LTB4 with increasing concentrations of extracellular calcium (Figure 1E). Correlating with calcium influx, CGD neutrophils produced more LTB4, particularly at lower extracellular calcium concentrations (Figure 1E), and differences in LTB4 made by WT and CGD neutrophils were overcome by providing more extracellular calcium. Taken together, these results show that extracellular calcium affects LTB4 production, and enhanced calcium influx in the absence of NADPH oxidase contributes to greater LTB4 production by zymosan-activated CGD neutrophils.
Higher levels of LTB4 produced by zymosan-activated CGD PMNs was dependent on a LTB4-BLT1 feedback loop
LTB4 production can be amplified by cross talk between neutrophils, as LTB4 bound to BLT1 can stimulate calcium entry to drive additional LTB4 synthesis.13,29 We investigated whether an LTB4-BLT1 feedback loop also contributed to the differential production of LTB4 observed between zymosan-activated WT and CGD neutrophils. In contrast to studies of neutrophils at a density of 4 × 106 cells/mL (Figure 1A), LTB4 levels were similar for CGD and WT neutrophils when stimulated at fourfold lower cell density for 1 hour (Figure 2A). However, after the duration of incubation was increased to 5 hours, LTB4 from CGD neutrophils rose to significantly higher levels (Figure 2A). The increased amount of LTB4 was not accounted for by differences in WT and CGD neutrophils undergoing apoptosis (9.4% ± 1.4% and 20.9% ± 1.4%; n = 3, respectively, after 5 hours). Thus, the increased amount of LTB4 recovered from CGD neutrophils was cell density and time dependent, suggesting that a paracrine feedback loop augments LTB4 production by CGD neutrophils. Providing further support for such a loop, the BLT1 antagonist U75302 significantly reduced LTB4 produced by CGD but not WT neutrophils at a cell density of 4 × 106/mL (Figure 2B). CGD PMNs stimulated for 5 hours at a lower cell density also produced significantly less LTB4 when BLT1 was antagonized (Figure 2B). Finally, LTB4 induced a rapid influx of calcium that was more sustained in CGD neutrophils compared with WT, resulting in significantly higher intracellular calcium levels in CGD over a range of extracellular calcium concentrations (Figure 2C-D). Collectively, these results show that cross talk between LTB4 and its BLT1 receptor played an important role in augmenting LTB4 generation by zymosan-activated CGD neutrophils, and implicate increased LTB4-induced calcium influx in CGD neutrophils as an amplifier of this cross talk.
![LTB4 production in CGD PMN was dependent on cell density and on the LTB4 receptor BLT1. (A) Mouse BM neutrophils (1 × 106/mL) were stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+). n = 18 per group (1 hour) and n = 8 per group (5 hours) from more than 3 separate experiments. #P < .05; ##P < .01; ###P < .001, by paired t test; ***P < .001, by Student t test. (B) Mouse BM neutrophils (4 × 106/mL or 1 × 106/mL) were pretreated with 10 µM U75302 for 10 minutes and then stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 10 µM U75302. n = 4 per group from 2 separate experiments. **P < .01, paired t test. (A-B) LTB4 levels in culture supernatant were measured by ELISA. Data are means ± standard error of the mean. (C) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils loaded with 3 µM indo-1 were stimulated with 0.5 ng/mL LTB4 with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Data are means ± standard error of the mean from 2 to 3 independent experiments. *P < .05, by Student t test. MOI, multiplicity of infection.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/12/10.1182_blood.2019003525/4/m_bloodbld2019003525f2.png?Expires=1769089294&Signature=SH8S1-vfOHOPEtg6dZpiQGsB771L7T3XQeLGquevEfYSJJ9p7j2nzQM1ENYGAdSkVuVdE~F4cKcUUDVQzIF3zq96V8~4YpGGHK~eKcXNFOq0ObAsasSFt9UnUBrZ6654EDsnec0SerHC3jt1hH~vzWbpLBSmVPaYV5FZfWhNwxTLKQUnknm~GSH2LHic-Tz-pvU3c70Uifq9DFS5Mxwzlu8j8QzdYZeYpy2GHWUktJuCATE7lbPaay22Ii22E2DHGIGpxP9TD4gORu3mdE6h8hH8Rb8miGb2fIniUCG2c~O3piD6lSVxQe9JE07LcIXElFJuM3pwWUOy084wLSJTeg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
LTB4 production in CGD PMN was dependent on cell density and on the LTB4 receptor BLT1. (A) Mouse BM neutrophils (1 × 106/mL) were stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+). n = 18 per group (1 hour) and n = 8 per group (5 hours) from more than 3 separate experiments. #P < .05; ##P < .01; ###P < .001, by paired t test; ***P < .001, by Student t test. (B) Mouse BM neutrophils (4 × 106/mL or 1 × 106/mL) were pretreated with 10 µM U75302 for 10 minutes and then stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 10 µM U75302. n = 4 per group from 2 separate experiments. **P < .01, paired t test. (A-B) LTB4 levels in culture supernatant were measured by ELISA. Data are means ± standard error of the mean. (C) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils loaded with 3 µM indo-1 were stimulated with 0.5 ng/mL LTB4 with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Data are means ± standard error of the mean from 2 to 3 independent experiments. *P < .05, by Student t test. MOI, multiplicity of infection.
LTB4 production in CGD PMN was dependent on cell density and on the LTB4 receptor BLT1. (A) Mouse BM neutrophils (1 × 106/mL) were stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+). n = 18 per group (1 hour) and n = 8 per group (5 hours) from more than 3 separate experiments. #P < .05; ##P < .01; ###P < .001, by paired t test; ***P < .001, by Student t test. (B) Mouse BM neutrophils (4 × 106/mL or 1 × 106/mL) were pretreated with 10 µM U75302 for 10 minutes and then stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 10 µM U75302. n = 4 per group from 2 separate experiments. **P < .01, paired t test. (A-B) LTB4 levels in culture supernatant were measured by ELISA. Data are means ± standard error of the mean. (C) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils loaded with 3 µM indo-1 were stimulated with 0.5 ng/mL LTB4 with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Data are means ± standard error of the mean from 2 to 3 independent experiments. *P < .05, by Student t test. MOI, multiplicity of infection.
LTB4 production in immune-complex–activated neutrophils was increased in the absence of NADPH oxidase activity
To evaluate whether enhanced LTB4 production by CGD PMNs could be triggered by another receptor that activates calcium influx and NADPH oxidase activity,32 we studied responses to FcγR stimulation by plate-bound immune complexes (ICs). Significantly higher levels of LTB4 were recovered from murine CGD neutrophils or DPI-treated human neutrophils after IC stimulation for 1 hour at a cell density of 4 × 106/mL (supplemental Figure 2A-B). LTB4 levels from IC-stimulated WT murine PMNs were unchanged by the addition of SOD and catalase (supplemental Figure 2C). The increased generation of LTB4 by IC-activated CGD neutrophils was dependent on cell density and the BLT1 receptor (supplemental Figure 2D-E). Hence, results for IC stimulation were similar to those obtained for zymosan-activated LTB4 production.
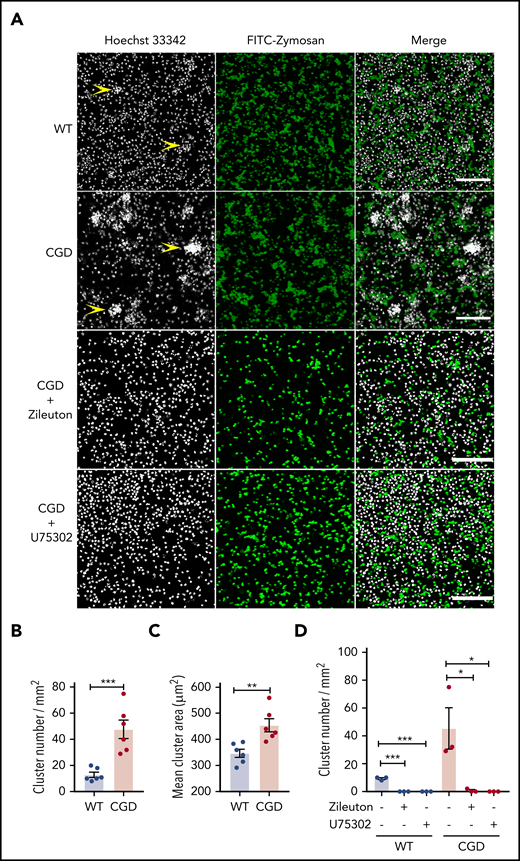
CGD neutrophils spontaneously formed larger and increased numbers of clusters than WT cells in the presence of zymosan in vitro, and cluster formation was LTB4 and BLT1 dependent
As both a chemoattractant and trigger for additional LTB4 production, LTB4 can mediate the formation of neutrophil clusters, both in vivo16 and in vitro.37,38 We examined whether zymosan-stimulated LTB4 formation could enhance CGD neutrophil aggregation in vitro. When incubated with zymosan, neutrophils aggregated into clusters, which were present in greater numbers and larger size for CGD compared with WT cells (Figure 3A-C). Cluster formation was abrogated by the 5-LOX inhibitor zileuton or the BLT1 antagonist U75302 (Figure 3A,D). Neither WT nor CGD neutrophils formed clusters after 1 hour in media alone (data not shown).Taken together with results described in Figures 1 and 2, these findings suggest that increased LTB4 production by zymosan-activated CGD neutrophils leads to the formation of more numerous and larger clusters of neutrophils via cross talk involving LTB4 and BLT1.
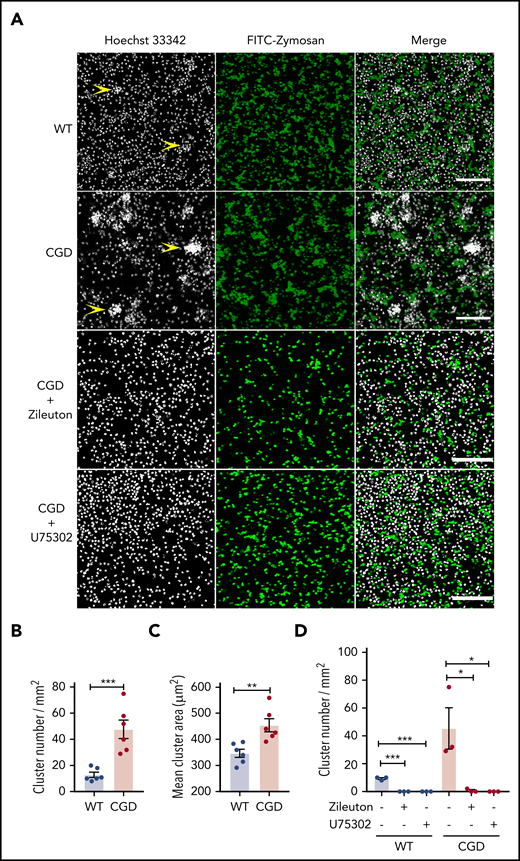
CGD neutrophils spontaneously formed larger and increased amounts of clusters in the presence of zymosan in vitro. Mouse BM neutrophils (4 × 106/mL) stained with 5 µg/mL Hoechst 33342 were stimulated for 1 hour with FITC-zymosan (MOI = 2). Images were acquired with LASX software (Leica Microsystems). (A) Representative images showing the neutrophil nuclei (white) and FITC-zymosan (green) and merged images of the 2 channels acquired by confocal microscopy. Examples of clusters are indicated by yellow arrowheads. Scale bars, 100 µm. (B) Quantification of the clusters and (C) cluster area. n = 6 in each group from 3 separate experiments. (D) CGD mouse BM neutrophils (4 × 106/mL) stained with 5 µg/mL Hoechst 33342 were preincubated with 30 µM zileuton or 10 µM U75302 for 10 minutes and then stimulated for 1 hour with FITC-zymosan (MOI = 2) in the presence of 30 µM zileuton or 10 µM U75302. Images were acquired by confocal microscope. (E) Quantification of the cluster number. n = 3 in each group from 2 separate experiments. Data are means ± standard error of the mean. *P < .05, **P < .01; ***P < .001, by Student t test. MOI, multiplicity of infection.
CGD neutrophils spontaneously formed larger and increased amounts of clusters in the presence of zymosan in vitro. Mouse BM neutrophils (4 × 106/mL) stained with 5 µg/mL Hoechst 33342 were stimulated for 1 hour with FITC-zymosan (MOI = 2). Images were acquired with LASX software (Leica Microsystems). (A) Representative images showing the neutrophil nuclei (white) and FITC-zymosan (green) and merged images of the 2 channels acquired by confocal microscopy. Examples of clusters are indicated by yellow arrowheads. Scale bars, 100 µm. (B) Quantification of the clusters and (C) cluster area. n = 6 in each group from 3 separate experiments. (D) CGD mouse BM neutrophils (4 × 106/mL) stained with 5 µg/mL Hoechst 33342 were preincubated with 30 µM zileuton or 10 µM U75302 for 10 minutes and then stimulated for 1 hour with FITC-zymosan (MOI = 2) in the presence of 30 µM zileuton or 10 µM U75302. Images were acquired by confocal microscope. (E) Quantification of the cluster number. n = 3 in each group from 2 separate experiments. Data are means ± standard error of the mean. *P < .05, **P < .01; ***P < .001, by Student t test. MOI, multiplicity of infection.
In the lung, CGD mice exhibited enhanced zymosan-induced neutrophil infiltration, which was associated with increased LTB4
We next investigated whether LTB4 played a role in the amplified neutrophil response of CGD mice to zymosan in vivo. Sterile fungal cell walls induced increased acute neutrophilic inflammation in CGD compared with WT mice, which progresses to chronic pyogranulomatous lesions.2,3 After instillation of zymosan into the lung, bronchoalveolar lavage (BAL) and lung neutrophils increased in both genotypes but were significantly higher in CGD within 8 to 14 hours (Figure 4A-B). The increase in neutrophils plateaued in the WT mice by 8 hours, but continued in CGD mice, so that differences between genotypes were even more substantial after 24 hours (Figure 4A-B). As early as 8 hours after the zymosan challenge, foci of neutrophils were visible in the lung parenchyma in both WT and CGD (Figure 4C). Although there was no difference in the number of foci (Figure 4D), their size was significantly greater in CGD mouse lungs compared with WT lungs (Figure 4E). Over the next 16 hours, neutrophil foci further increased in size in CGD mice, but became less visible in WT mice (supplemental Figure 3A). The increased neutrophilic inflammation induced by zymosan in CGD was accompanied by significantly elevated levels of LTB4 in the lung and BAL (Figure 4F-G).
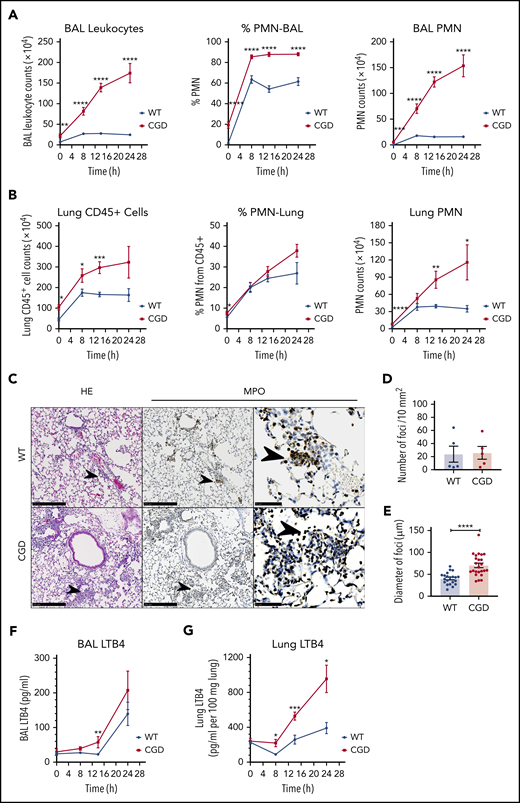
Zymosan-induced lung inflammation was greater in CGD mice and was associated with increased LTB4. WT and CGD mice were challenged with 20 µg IN zymosan. BAL and lung tissues were collected at different time points. (A) Total leukocyte counts from 3 mL BAL fluid. The percentage of PMNs was determined by Cytospin with Wright-Giemsa stain. BAL PMN counts were calculated by Cytospin results. n ≥ 13 in each group from more than 3 independent experiments. (B) Lung cell counts from the right lung inferior lobe. The percentage of CD45+ cells and PMNs (CD45+Ly6G+CD11b+) was determined by flow cytometry. CD45+ cell and PMN counts were calculated by flow cytometry. n ≥ 3 in each group from more than 3 separate experiments. (C) Formalin-fixed, paraffin-embedded lung sections were stained with hematoxylin and eosin or processed for immunohistochemical study with MPO. Scale bars: 250 µm (left and middle panels); 50 µm (right panel). (D) Quantification of foci and (E) diameter of foci obtained with immunohistochemical staining with MPO of slides of cells from 5 different mice in each group. LTB4 levels in the first milliliter of BAL fluid supernatant (F) and whole-lung homogenate supernatant (G) were measured by ELISA. (F) n ≥ 7 in each group from more than 3 separate experiments; (G) n ≥ 3 in each group from more than 2 separate experiments. (A-B,E-G) Data are means ± standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001, by Student t test.
Zymosan-induced lung inflammation was greater in CGD mice and was associated with increased LTB4. WT and CGD mice were challenged with 20 µg IN zymosan. BAL and lung tissues were collected at different time points. (A) Total leukocyte counts from 3 mL BAL fluid. The percentage of PMNs was determined by Cytospin with Wright-Giemsa stain. BAL PMN counts were calculated by Cytospin results. n ≥ 13 in each group from more than 3 independent experiments. (B) Lung cell counts from the right lung inferior lobe. The percentage of CD45+ cells and PMNs (CD45+Ly6G+CD11b+) was determined by flow cytometry. CD45+ cell and PMN counts were calculated by flow cytometry. n ≥ 3 in each group from more than 3 separate experiments. (C) Formalin-fixed, paraffin-embedded lung sections were stained with hematoxylin and eosin or processed for immunohistochemical study with MPO. Scale bars: 250 µm (left and middle panels); 50 µm (right panel). (D) Quantification of foci and (E) diameter of foci obtained with immunohistochemical staining with MPO of slides of cells from 5 different mice in each group. LTB4 levels in the first milliliter of BAL fluid supernatant (F) and whole-lung homogenate supernatant (G) were measured by ELISA. (F) n ≥ 7 in each group from more than 3 separate experiments; (G) n ≥ 3 in each group from more than 2 separate experiments. (A-B,E-G) Data are means ± standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001, by Student t test.
Neutrophils were the major source of LTB4 in zymosan-induced lung inflammation in CGD
Although neutrophils are an important source of LTB4, this leukotriene can also be synthesized by other cells.21,22 We therefore used an anti-Ly6G antibody to assess LTB4 levels after neutrophil depletion.35 Neutrophils in the peripheral blood, BAL, and lung were significantly decreased 14 hours after zymosan challenge in both WT and CGD mice that received anti-Ly6G compared with zymosan-challenged mice receiving isotype control antibody (Figure 5A-B,D). LTB4 levels in lung tissue homogenates were significantly reduced in neutrophil-depleted CGD mice (Figure 5E), and BAL also showed a trend for decreased LTB4 (Figure 5C). Furthermore, there was no difference in the fraction of apoptotic neutrophils in WT and CGD mouse lungs 14 hours after zymosan challenge (17.3% ± 3.0% and 17.4% ± 5.4%; n = 3, respectively), indicating that increased LTB4 recovered from CGD mouse lungs did not reflect reduced neutrophil apoptosis. Collectively, these results indicate that neutrophils were the major source of increased LTB4 in CGD mouse lungs during zymosan-induced inflammation.
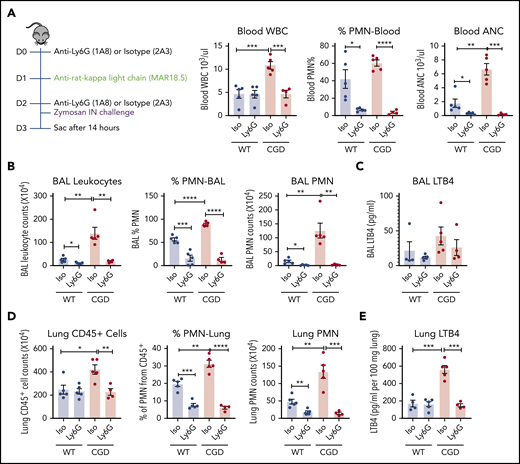
PMNs were the major source of LTB4 in zymosan-induced lung inflammation. Mice were sequentially injected intraperitoneally with anti-Ly6G/isotype and anti-rat κ light chain, followed by IN instillation of 20 µg zymosan. (A) After 14 hours, total WBCs, percentage of PMNs, and absolute neutrophil count (ANC) were determined in peripheral blood by complete blood count and differential. (B) Cell counts from 3 mL BAL. Cell composition was analyzed by Cytospin with Wright-Giemsa stain. (C) LTB4 levels in the first milliliter of BAL fluid supernatant was measured by ELISA. (D) Lung cell counts from the right lung inferior lobe. The percentage of PMN (CD45+Ly6CintCD11b+) was determined by flow cytometry. CD45+ cell and PMN counts were calculated by flow cytometry. (E) LTB4 levels in whole-lung homogenate supernatant was measured by ELISA. n = 4 in each group from 2 separate experiments. Data are means ± standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001, by Student t test.
PMNs were the major source of LTB4 in zymosan-induced lung inflammation. Mice were sequentially injected intraperitoneally with anti-Ly6G/isotype and anti-rat κ light chain, followed by IN instillation of 20 µg zymosan. (A) After 14 hours, total WBCs, percentage of PMNs, and absolute neutrophil count (ANC) were determined in peripheral blood by complete blood count and differential. (B) Cell counts from 3 mL BAL. Cell composition was analyzed by Cytospin with Wright-Giemsa stain. (C) LTB4 levels in the first milliliter of BAL fluid supernatant was measured by ELISA. (D) Lung cell counts from the right lung inferior lobe. The percentage of PMN (CD45+Ly6CintCD11b+) was determined by flow cytometry. CD45+ cell and PMN counts were calculated by flow cytometry. (E) LTB4 levels in whole-lung homogenate supernatant was measured by ELISA. n = 4 in each group from 2 separate experiments. Data are means ± standard error of the mean. *P < .05; **P < .01; ***P < .001; ****P < .0001, by Student t test.
Neutrophil recruitment in CGD mice in the initial response to zymosan is dependent on ongoing LTB4 synthesis
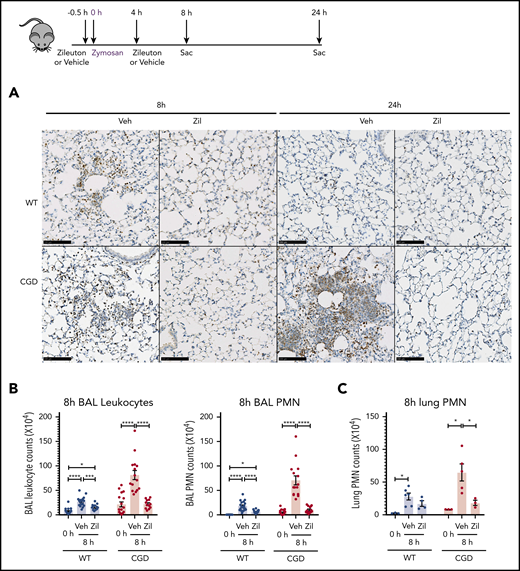
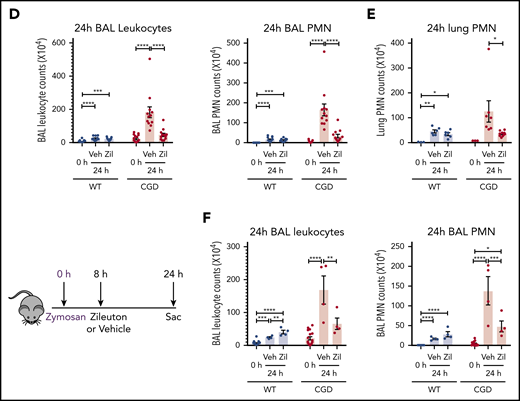
To determine whether the increased neutrophilic inflammation elicited by zymosan in CGD mice was mediated by LTB4, we administered zileuton, which has a half-life of ≈4 to 6 hours, just before zymosan and again 4 hours later.33 Zileuton treatment significantly reduced LTB4 levels in BAL and lung homogenates from WT and CGD mice at 8 and 24 hours after zymosan challenge (supplemental Figure 5A-D). At 8 hours, lung parenchyma showed a marked reduction in neutrophil foci in zileuton-treated mice of both genotypes (Figure 6A), associated with significantly reduced BAL neutrophils (Figure 6B). Total lung neutrophils were also significantly decreased in zileuton-treated CGD mice at 8 hours (Figure 6C). At 24 hours after zymosan challenge, neutrophil infiltrates seen on lung histology remained markedly reduced in zileuton–treated CGD mice (Figure 6A), associated with a significant decrease in BAL and lung neutrophils (Figure 6D-E). These results show that LTB4 is an important mediator of neutrophil recruitment to the lung for both WT and CGD mice in the first 8 hours of zymosan-induced inflammation. Moreover, in CGD mice, early inhibition of LTB4 synthesis had a prolonged effect, markedly reducing lung neutrophilic inflammation, even at 24 hours after zymosan challenge.

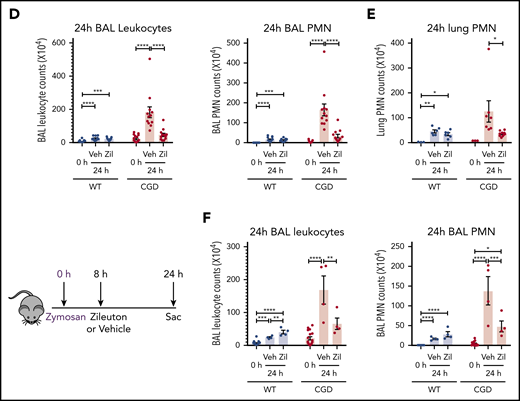
Increased neutrophilic inflammation in CGD mice in response to zymosan was dependent on LTB4. (A-E) Wild-type and CGD mice were treated with zileuton or vehicle 30 minutes before and 4 hours after 20 μg zymosan challenge and examined at 8 or 24 hours, as indicated. (A) Representative immunohistochemical images of lung sections stained with MPO. Scale bars, 100 µm. (B,D) BAL leukocytes and PMNs (identified by Cytospin) and (C,E) lung PMNs (CD45+Ly6G+CD11b+ by flow cytometry) were analyzed 8 hours or 24 hours after challenge. (F) Mice were treated with zileuton or vehicle 8 hours after 20 µg zymosan challenge. BAL leukocytes and PMNs (identified by Cytospin) were analyzed 24 hours after challenge. n ≥ 3 per group from at least 2 separate experiments. Data are means ± standard error of the mean . (B-F) *P < .05, **P < .01, ***P < .001, ****P < .0001, by 1-way ANOVA.
Increased neutrophilic inflammation in CGD mice in response to zymosan was dependent on LTB4. (A-E) Wild-type and CGD mice were treated with zileuton or vehicle 30 minutes before and 4 hours after 20 μg zymosan challenge and examined at 8 or 24 hours, as indicated. (A) Representative immunohistochemical images of lung sections stained with MPO. Scale bars, 100 µm. (B,D) BAL leukocytes and PMNs (identified by Cytospin) and (C,E) lung PMNs (CD45+Ly6G+CD11b+ by flow cytometry) were analyzed 8 hours or 24 hours after challenge. (F) Mice were treated with zileuton or vehicle 8 hours after 20 µg zymosan challenge. BAL leukocytes and PMNs (identified by Cytospin) were analyzed 24 hours after challenge. n ≥ 3 per group from at least 2 separate experiments. Data are means ± standard error of the mean . (B-F) *P < .05, **P < .01, ***P < .001, ****P < .0001, by 1-way ANOVA.
To further define the temporal requirements for LTB4 synthesis in zymosan-elicited neutrophil recruitment, we delayed zileuton administration until 8 hours after zymosan challenge. BAL leukocytes and neutrophils remained significantly reduced in CGD mice at 24 hours after zymosan to counts similar to those in WT mice (Figure 6F). There was also a substantial reduction in lung neutrophils, detected by histology and flow cytometry analysis (supplemental Figure 5E-F). However, delaying zileuton administration to 24 hours after zymosan did not reduce BAL neutrophils in CGD mice at 48 hours after zymosan, and parenchymal infiltrates continued to increase in size (supplemental Figure 6A-D). In addition, although inflammation was suppressed for 24 hours in CGD mice with early zileuton treatment, inflammation then progressed, even if additional zileuton was given (data not shown). This result indicates that other proinflammatory agents were responsible for the inflammation in CGD mice after 24 hours. In aggregate, these results demonstrate that LTB4 synthesis was required to mediate neutrophil recruitment in both WT and CGD mice during the first 8 hours of the response to zymosan. Moreover, the progressive inflammation in CGD mice between 8 and 24 hours was driven by ongoing LTB4 production.
IL-1β, CXCL2, and G-CSF levels in WT and CGD mice were similar in the first 8 hours and were not reduced by zileuton
Fungal cell walls are potent triggers for the NF-κB-mediated production of proinflammatory cytokines that promote neutrophil recruitment in the host response.39,40 BAL levels of IL-1β, G-CSF, and CXCL2 were all increased by 8 hours after zymosan challenge compared with naïve mice (supplemental Figure 7A,C,E). However, there was no difference between genotypes, although neutrophils were already significantly increased in CGD mouse lungs (Figure 4). Over the next 16 hours, IL-1β, CXCL2, and G-CSF levels were higher in CGD BAL samples compared with those from WT samples (supplemental Figure 7), correlating with the progressive increase in inflammation in CGD mice. Of note, early treatment with zileuton did not reduce BAL cytokine levels in zymosan-challenged mice, at either 8 or at 24 hours (supplemental Figure 7B,D,F), while profoundly reducing lung neutrophils in CGD mice (Figure 6). Thus, BAL IL-1β, CXCL2, and G-CSF did not correlate with neutrophil recruitment to the lungs of CGD mice in the first 24 hours, indicating that LTB4 played a dominant and nonredundant role in driving the increased neutrophilic inflammation.
The LTB4 receptor BLT1 was required for amplified neutrophil recruitment in zymosan-induced lung inflammation in CGD
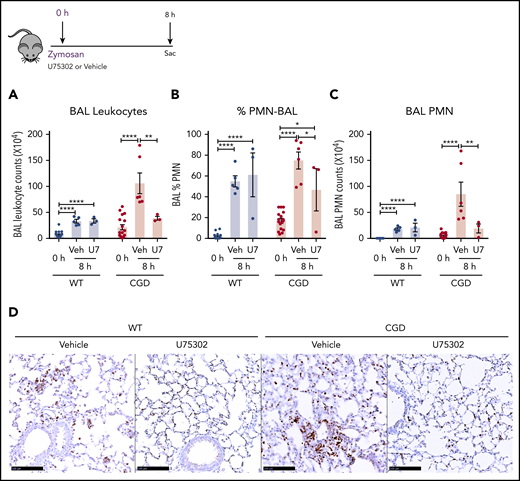
Based on our in vitro experiments, we hypothesized that elevated LTB4 production and neutrophil recruitment to the lungs of zymosan-challenged CGD mice was augmented by cross talk with BLT1 receptors. Therefore, we examined the effect of antagonizing BLT1 with U75302, administered at the time of zymosan challenge. BAL neutrophils and lung neutrophil foci were significantly decreased by U75302 in CGD mice at 8 hours after challenge, whereas these were unaffected in WT mice (Figure 7A-D). These results suggest that cross talk between LTB4 and its BLT1 receptor in CGD mice is essential for amplifying early neutrophil recruitment during zymosan-induced lung inflammation.
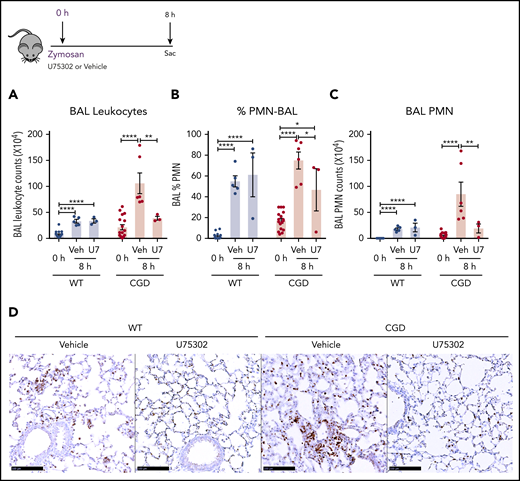
The LTB4 receptor BLT1 was essential for amplified neutrophilic lung inflammation in zymosan-challenged CGD mice. WT and CGD mice were challenged with 20 µg zymosan and either U75302 or vehicle. BAL leukocytes (A) and PMNs (B-C), identified by Cytospin, were analyzed 24 hours after challenge. (D) Representative images of lung sections stained with MPO. Scale bars, 100 μm. n ≥ 3 per group from at least 2 separate experiments. Data are means ± standard error of the mean . *P < .05; **P < .01; ***P < .001; ****P < .0001, by 1-way ANOVA.
The LTB4 receptor BLT1 was essential for amplified neutrophilic lung inflammation in zymosan-challenged CGD mice. WT and CGD mice were challenged with 20 µg zymosan and either U75302 or vehicle. BAL leukocytes (A) and PMNs (B-C), identified by Cytospin, were analyzed 24 hours after challenge. (D) Representative images of lung sections stained with MPO. Scale bars, 100 μm. n ≥ 3 per group from at least 2 separate experiments. Data are means ± standard error of the mean . *P < .05; **P < .01; ***P < .001; ****P < .0001, by 1-way ANOVA.
Discussion
The leukocyte NADPH oxidase is important, not only for killing microbes, but also for modulating inflammation.1 CGD has a well-known association with neutrophilic hyperinflammation,1-4,7,41,42 yet key mediators dysregulated by the absence of NADPH oxidase ROS are incompletely defined. In this study, we established a link between increased calcium influx in activated CGD neutrophils and elevated LTB4 production that was also dependent on amplified cross talk with its receptor BLT1. In vivo, elevated and prolonged LTB4 production in CGD mice drove increased and progressive pulmonary neutrophil infiltration in zymosan-induced lung inflammation.
Our results confirm and extend prior studies25,26,43 showing that activated CGD neutrophils have relative calcium overload and establish increased LTB4 generation as a functional consequence. Prior studies compared LTB4 production by normal and CGD neutrophils, but only in response to calcium ionophores, rather than physiologic stimuli, and with variable results.44-46 In our study, we showed that NADPH oxidase-deficient mouse and human neutrophils activated with either zymosan or immune complexes produced more LTB4, which was related to 2 factors. First, higher LTB4 production in CGD PMNs depended on increased calcium influx in the absence of electrogenic activity of the NAPDH oxidase. Second, increased LTB4 production by CGD was dependent on paracrine cross talk with BLT1 and associated with higher intracellular calcium levels elicited by LTB4. Our results are also consistent with those of a study showing that human CGD neutrophils stimulated with fMLF produced more arachidonic acid, the precursor to LTB4, compared with normal neutrophils.26
ROS-mediated degradation did not appear to be a major factor in the much lower levels of LTB4 recovered from WT PMNs. LTB4 chemotactic activity is rapidly decreased by hydroxyl radicals in cell-free systems,47 and it was proposed that NADPH oxidase ROS can also degrade leukotrienes in normal neutrophils. However, 1 study focused on LTC4,44 and there was little effect of oxidant scavengers on recovery of LTB4 in another report.45
Aspergillus pneumonia in CGD is associated with pyogranulomatous inflammation and abscess formation,48-50 and sterile fungal cell walls induce a similar pathology in mouse models of CGD.2,3 We used zymosan-induced lung inflammation to probe the role of LTB4 in the aberrant inflammatory response to fungal cell walls in CGD. LTB4 mediated neutrophil recruitment in both WT and CGD mice during the first 8 hours and was responsible for the markedly increased neutrophilic inflammation in CGD mice during the first 24 hours of the response. Neutrophils were the major source of LTB4. LTB4 is recognized as an important early mediator of neutrophil recruitment during fungal infection51-55 and also caused pulmonary capillaritis in a model of candidemia.56 Although normally beneficial to the host response to fungal infection, overexuberant LTB4-mediated neutrophil recruitment could contribute to the neutrophilic inflammation and abscess formation characteristic of Aspergillus pneumonia in CGD and further impair its control. In cutaneous inflammation elicited by sterile A. fumigatus hyphae, CGD mice also had excessive acute inflammation associated with elevated LTB4 levels.4 In addition, zileuton reduced the enhanced thioglycolate-induced peritonitis in p47phox−/− CGD mice,42 suggesting that LTB4 may be an important driver of neutrophilic inflammation in CGD in other inflammatory settings.
Feed-forward amplification of local LTB4 production leading to rapid focal accumulation of neutrophils is well known,13-17 as LTB4 acts on neutrophils, both as a chemoattractant and to activate additional LTB4 synthesis. We showed that CGD neutrophils incubated with zymosan in vitro spontaneously formed larger and more numerous clusters than WT neutrophils, and cluster formation was dependent on LTB4 and its BLT1 receptor. Although the dynamics of neutrophil clustering in the lungs of CGD mice after zymosan needs further investigation, these in vitro observations are consistent with the pattern of zymosan-induced neutrophil accumulation in the lung. By 8 hours after challenge, neutrophil foci were evident in both WT and CGD lung parenchyma, but already larger in size in CGD, and their formation was dependent on LTB4 and BLT1. Thus, we propose that higher local production of LTB4 by CGD neutrophils amplified focal neutrophil recruitment in response to fungal cell walls.
LTB4 is an early mediator of neutrophil recruitment in many noninfectious inflammatory settings, including inflammatory arthritis and allergic asthma, and initiates a temporal lipid-cytokine-chemokine cascade.11,14,15,57 Neutrophilic hyperinflammation in CGD is associated with increased proinflammatory cytokines and chemokines produced by myeloid cells, reflecting in part increased activation of NF-κB and of the IL-1 axis.1-3,6,7,41 However, we found no correlation of IL-1β, G-CSF, and CXCL2 levels with the marked differences in early neutrophil infiltration in CGD and WT mice after zymosan, and their levels remained high even when CGD inflammation was suppressed by zileuton. Thus, neutrophil recruitment appears to be independent of these cytokines in the initial phase of the zymosan response. However, progressive inflammation in CGD mice was independent of LTB4, indicating that contributions from other inflammatory mediators become important.
NADPH oxidase deficiency has pleiotropic effects on modulating cellular responses to immune activation.1 The importance of LTB4 as the initial driver of amplified zymosan-induced inflammation in CGD differs from the response to endogenous danger-associated molecular patterns in a peritoneal injury model.41 In the latter, increased IL-1α release upon danger-associated molecular pattern activation of sentinel hematopoietic cells, likely macrophages, led to a rapid increase in G-CSF levels in CGD mice. In turn, elevated G-CSF enhanced mobilization of marrow neutrophils that drove increased neutrophilic infiltration. These differences illustrate how aberrant inflammation in CGD can be initiated by dysregulation of different myeloid cells and pathways, depending on the setting.
Our study provides an important new view for investigating the clinical use of inhibitors of LTB4 synthesis22,58 or BLT1 function59 in the treatment of CGD patients. Although there are some differences between mouse and human immunology,60 evidence supports an important role for LTB4 in directing neutrophil recruitment in humans and nonhuman primates, including in pulmonary inflammation.61-64 LTB4 also mediates human neutrophil swarming in vitro.38 Our results also implicate intracellular calcium overload as a factor in the amplified LTB4 production by NAPDH oxidase-deficient neutrophils. Neutrophil deletion of STIM1 and/or STIM2, integral components of the SOCE pathway, is protective in different inflammation models.32,65 CRAC channel inhibitors or inhibition of other channels that regulate calcium influx have been suggested for treatment of inflammatory diseases23,29,66-68 and could also be considered as an approach to ameliorate CGD hyperinflammation.
In summary, study demonstrates that NADPH oxidase limits LTB4 production by acting on calcium entry and that increased production of LTB4 can promote overexuberant neutrophil recruitment in CGD. It provides new insights into how NADPH oxidase can constrain production of proinflammatory mediators, which in this case is independent of direct effects of ROS, and how limiting calcium influx and LTB4 production play an important role in tuning neutrophil recruitment.
For more detailed protocols and/or original data, please contact the corresponding author by e-mail.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Malewaan Kitcharoensakkul (Department of Pediatrics, Washington University School of Medicine) for referring the CGD patient for neutrophil studies, and Tina McGrath and Becky Kolb for assistance with manuscript preparation.
This work was supported by grants from the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (R01HL140837), NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR072212), the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (M.C.D.), and by an NIH grant from the National Institute on Aging (1K08AI119134) (R.A.C). This work was also supported by the Hope Center Alafi Neuroimaging Laboratory and by a NIH Shared Instrumentation grant (S10 RR027552).
Authorship
Contribution: Z.S., R.A.C., C.J.L., and M.C.D. designed research; Z.S., G.H., D.G., L.C.P., J.G., C.J.L., and R.A.C performed and/or analyzed the results of the experiments; Z.S. and R.A.C prepared the figures; and Z.S. and M.C.D wrote the manuscript with input from L.C.P., C.J.L., and R.A.C.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mary C. Dinauer, Washington University School of Medicine in St. Louis, 660 S Euclid Ave, PO Box 8208, St. Louis, MO 63110; e-mail: mdinauer@wustl.edu.

![CGD mouse neutrophils had increased LTB4 production and elevated calcium influx compared with WT, and oxidant scavengers did not increase LTB4 in WT neutrophils. (A) Mouse BM neutrophils (4 × 106/mL) were stimulated for various times with zymosan (MOI = 2 zymosan: 1 PMN) in RPMI 1640 (0.4 mM Ca2+; n ≥ 3). *P < .05; ****P < .0001), by Student t test. (B) Mouse BM neutrophils (4 × 106/mL) were pretreated with 200 U/mL SOD and 200 U/mL catalase for 10 minutes and then stimulated for 1 hour with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 200 U/mL SOD and 200 U/mL catalase. n = 4 per group from 2 separate experiments. *P < .05, by paired t test. NS, not significant. (C-D) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils (1 × 106/mL) loaded with 3 µM indo-1 were stimulated with (C) Zymosan (100 µg/mL; MOI ≈ 10) with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Mean ± standard error of the mean from 2 to 4 independent experiments. *P < .05; **P < .01, by Student t test. (E) Mouse BM neutrophils (4 × 106/mL) were stimulated for 1 hour with zymosan (MOI = 2) in Hanks’ balanced salt solution with the indicated amounts of extracellular calcium. LTB4 levels in culture supernatant was measured by ELISA. n ≥ 3 per group from 3 separate experiments. Data are means ± SEM. *P < .01; ***P < .001, by Student t test. MOI, multiplicity of infection.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/12/10.1182_blood.2019003525/4/m_bloodbld2019003525f1.png?Expires=1769175178&Signature=eqE0N5xu-SUD0OuNfsYRvNIwST8YES~Fm-pf5HDRTn0TOHIUD83cJIPs-hfnSutRDya9NmptrOSN06qeBgsrW7edZ~voU7m0y~N-jXoSuES2563a69LKYHTVSYMTa3XruKo-JgqOnu~mYWlAN8gr9t56kFBwkRVsVrqSXZmByorh-cL2rgI~v7NQPmoFcRhzVyMDx2lPQjHg5jgcDIXkLfebTGOe5W~YfSuKa~LHxG9nz2c5ONuP81rxxwl3PtUPz1XvqKPD8YUiMRb6CRhkFomd-nErXfg42SBD4DrB-VcSKf-cyOhGHGETwOoCLKDAZajoLnN~ppuuc2Gmt2oewg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![LTB4 production in CGD PMN was dependent on cell density and on the LTB4 receptor BLT1. (A) Mouse BM neutrophils (1 × 106/mL) were stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+). n = 18 per group (1 hour) and n = 8 per group (5 hours) from more than 3 separate experiments. #P < .05; ##P < .01; ###P < .001, by paired t test; ***P < .001, by Student t test. (B) Mouse BM neutrophils (4 × 106/mL or 1 × 106/mL) were pretreated with 10 µM U75302 for 10 minutes and then stimulated for 1 hour or 5 hours with zymosan (MOI = 2) in RPMI 1640 (0.4 mM Ca2+) in the presence of 10 µM U75302. n = 4 per group from 2 separate experiments. **P < .01, paired t test. (A-B) LTB4 levels in culture supernatant were measured by ELISA. Data are means ± standard error of the mean. (C) SOCE in neutrophils from WT and CGD mice was measured by flow cytometry. Mouse BM neutrophils loaded with 3 µM indo-1 were stimulated with 0.5 ng/mL LTB4 with the indicated amounts of extracellular calcium. (D) Quantification of SOCE (area under the curve [AUC]). Data are means ± standard error of the mean from 2 to 3 independent experiments. *P < .05, by Student t test. MOI, multiplicity of infection.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/12/10.1182_blood.2019003525/4/m_bloodbld2019003525f2.png?Expires=1769175178&Signature=e7DmKqRTO~9Wnq7DsDrpbFVYCIpbpTZugbCU7GKbtr57ZcuvJ4XLekUavmIKq1isLPBXKlj-oELVtpFMpLK1KmdTUQABEtMC7aOvA5OeAbLdjr2pkCt~44C1DH~Wu3wJSuljeVGs~HAY78d-3w6rZAdrJkAQVwic~1vZrxRLSVmCfnjLm8sT0GLAa2FU-BwtuBXjAgHOAMe8NluHFILEr39mspPnF5B20aCn8ZW9IJ4Pu7j3~7KOGnBuy-Wmeji-SS9tzgT8aLpQeOZuYSi6J5sr0m5MynHZNL8DK3Car6TylMpXLNcBq3JCKtxQ769lsGmjQxC2Lya9Cp~v2z0-Og__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)