Key Points
NT5C2 mutations are present in 16% of B-cell precursor ALL relapses and are clonal in one-third and subclonal in two-thirds of cases.
Subclonal NT5C2 mutations independently predict inferior outcome after relapse, although they are rapidly eradicated by relapse therapy.

Abstract
Activating mutations in cytosolic 5′-nucleotidase II (NT5C2) are considered to drive relapse formation in acute lymphoblastic leukemia (ALL) by conferring purine analog resistance. To examine the clinical effects of NT5C2 mutations in relapsed ALL, we analyzed NT5C2 in 455 relapsed B-cell precursor ALL patients treated within the ALL-REZ BFM 2002 relapse trial using sequencing and sensitive allele-specific real-time polymerase chain reaction. We detected 110 NT5C2 mutations in 75 (16.5%) of 455 B-cell precursor ALL relapses. Two-thirds of relapses harbored subclonal mutations and only one-third harbored clonal mutations. Event-free survival after relapse was inferior in patients with relapses with clonal and subclonal NT5C2 mutations compared with those without (19% and 25% vs 53%, P < .001). However, subclonal, but not clonal, NT5C2 mutations were associated with reduced event-free survival in multivariable analysis (hazard ratio, 1.89; 95% confidence interval, 1.28-2.69; P = .001) and with an increased rate of nonresponse to relapse treatment (subclonal 32%, clonal 12%, wild type 9%, P < .001). Nevertheless, 27 (82%) of 33 subclonal NT5C2 mutations became undetectable at the time of nonresponse or second relapse, and in 10 (71%) of 14 patients subclonal NT5C2 mutations were undetectable already after relapse induction treatment. These results show that subclonal NT5C2 mutations define relapses associated with high risk of treatment failure in patients and at the same time emphasize that their role in outcome is complex and goes beyond mutant NT5C2 acting as a targetable driver during relapse progression. Sensitive, prospective identification of NT5C2 mutations is warranted to improve the understanding and treatment of this aggressive ALL relapse subtype.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children.1 Despite an overall favorable prognosis, 15% to 20% of pediatric ALL patients suffer a relapse, which still is a leading cause of death from childhood cancer.2 Treatment of children with relapsed ALL is carried out with curative intent and commonly consists of intensified chemotherapy regimens and risk-adapted allocation to hematopoietic stem cell transplantation (HSCT).2 Event-free survival (EFS) rates after relapse are highly heterogeneous and range between 10% and 70% depending on clinical features and response to treatment of patients3-8 as well as genetics of the relapse leukemia cells.9-11 High-risk factors include an early recurrence of the disease after primary diagnosis,3-5 T-lineage immunophenotype,3-5 nonresponse or poor response to relapse therapy,6-8 mutation and/or deletion of the TP53 gene,9,10 and hypodiploidy.11 Further improvements in the treatment of relapsed ALL rely on the identification of novel predictive markers and treatment targets for support of individualized clinical decisions and targeted treatment strategies.
Recently, mutations in the cytosolic 5′-nucleotidase II (NT5C2) gene were suggested to drive resistance and relapse in ALL.12 By catalyzing the 5′ dephosphorylation of 6-hydroxy purine nucleoside monophosphates, NT5C2 participates in purine nucleotide metabolism and regulation of the intracellular nucleotide pool.13 NT5C2 mutations were identified selectively in relapsed ALL14-19 and were found to increase the enzymes activity.14,15,20-22 NT5C2 can act on metabolites of purine analogs,23 which are commonly used in maintenance therapy of ALL.24 Consistently, NT5C2 mutations have been identified preferentially in early relapses that occurred during or shortly after maintenance treatment.14,15,18 Leukemic cells expressing mutant NT5C2 showed resistance to thiopurine treatment in vitro14,15,19,21 and in vivo19 and had a selective growth advantage under 6-mercaptopurine treatment in a T-lineage ALL (T-ALL) mouse model.19 Overall, NT5C2 mutations have been detected in 19% to 38% of relapses of pediatric T-ALL15,17,18 and 3% to 45% of relapses of the much more frequently occurring pediatric B-cell precursor (BCP) ALL.14-16,21 However, all existing patient-directed studies on NT5C2 mutations were hampered by small cohorts not exceeding 71 (BCP-ALL) and 103 (T-ALL) relapsed patients as well as by heterogeneous relapse treatment.14-18,21 Therefore, reliable data on the response of NT5C2-mutant relapses to second-line treatment and the effect of NT5C2 mutations on survival of patients after relapse therapy is currently lacking. To examine the clinical effects of NT5C2 mutations and explore their potential as a novel marker and/or target in the treatment of relapsed ALL, we retrospectively studied 455 children with relapsed BCP-ALL from the ALL-REZ BFM 2002 trial cohort for mutations in NT5C2, assessed correlations with clinical parameters and outcome of patients, and tracked NT5C2-mutant clone development during and after relapse therapy.
Materials and methods
Patients
Between 2002 and 2012, the multicenter ALL-REZ BFM 2002 clinical trial (NCT00114348)25 enrolled 654 pediatric patients with first relapse of BCP-ALL in Germany. Written informed consent was obtained from patients or guardians. The treatment protocol was approved by the local medical research ethics committees. Patients with isolated extramedullary relapses were excluded due to scarce availability of extramedullary specimen (101 of 654). From the remaining 552 patients with isolated or combined bone marrow relapses, 455 patients (70%) were included in the present study based on successful DNA analyses. The 97 excluded patients with isolated or combined bone marrow relapses showed no selection bias with respect to the most relevant clinical factors (supplemental Table 1, available on the Blood Web site). The median follow-up time of relapsed patients in continuous complete remission was 11.6 years (range, 7.0-17.55 years).
Treatment of relapsed ALL according to the ALL-REZ BFM 2002 protocol included multiagent chemotherapy courses, repeated intrathecal administration of cytostatic drugs, and local irradiation if necessary.7,8,26 Intensification of treatment by allogeneic HSCT (allo-HSCT) was recommended for high-risk (S3/S4) relapsed patients once a second complete remission was achieved as well as for intermediate-risk (S2) relapsed patients with minimal residual disease levels ≥1E-03 after induction treatment (week 5).7,8,26 Complete remission was defined as a regenerating bone marrow with <5% leukemic blasts, peripheral blood without blasts and with evidence of regeneration, and absence of extramedullary leukemic involvement.7,8,26 Nonresponse was defined if the patient did not achieve complete remission after week 9 of relapse treatment (after induction and the first 2 courses of consolidation treatment).7,8,26 At initial diagnosis, patients of the present study were treated mainly according to protocols of the German ALL-BFM (68%) and COALL (26%) study groups by chemotherapy only. Only 2 relapsed patients had an allo-HSCT in first complete remission (ID 459 and ID 481).
Samples
All samples were enriched for mononuclear cells by Ficoll density gradient centrifugation prior to extraction of genomic DNA. For identification of NT5C2 mutations, bone marrow (n = 438), peripheral blood (n = 12), and testis (n = 5) samples taken at the time of first relapse were used (median percentage of leukemic blasts, 90%; range, 9% to 99%). Follow-up samples taken at the end of induction treatment, time of nonresponse, or second relapse were used to track NT5C2 mutations during and after relapse therapy. If a nonresponse sample from week 9 was not available, samples from bone marrow punctures before or after week 9 (end of week 4 to end of week 12) were used, provided the leukemic blast count was >30%.
NT5C2 sequence analysis
Amplicon-based next-generation sequencing (NGS) was carried out previously.27 The mean sequencing depth across the gene and between all samples was 970-fold. To detect true mutations against background error, we used a Bayesian approach as described previously.28 Because of variation in sequencing depth and mutation-specific sensitivity across the gene and between samples, we applied an uniform 2% variant allele frequency threshold for mutation calling. Details are given in the supplemental Methods. Sequencing by the method of Sanger was carried out using NT5C2 primers listed in supplemental Table 2. Sanger traces were analyzed by the Mutation Surveyor software version 5.0.0 (Softgenetics, State College, PA) with the GenBank sequence NG_042272.1 hg18/3829 as reference. For quantification of mutant alleles, the allelic peak ratio was calculated from traces using the Mutation Surveyor’s “simplified allele ratio” algorithm. Resequencing of all NGS-identified mutations by the method of Sanger revealed a good correlation between NGS variant allele frequency and Sanger allelic peak ratio (r2 = 0.87; supplemental Figure 1A). Therefore, we compiled variant allele frequencies and allelic peak ratios from NGS- and Sanger-detected mutations, respectively, and set a common cutoff point of 15% to separate subclonal from clonal mutations based on the distribution of the data (supplemental Figure 1B). All clonal mutations were confirmed as somatic mutations by paired samples from diagnosis or remission or second event being negative for the respective mutations by Sanger sequencing.
ASQ-PCR
For allele-specific quantitative real-time polymerase chain reaction (ASQ-PCR) of NT5C2-mutant variants, allele-specific primers with the 3′ terminal nucleotide matching the mutation plus additional mismatch on the second or the third base closest to the 3′ end were designed (supplemental Table 3)30 and evaluated following the guidelines for minimal residual disease analysis (illustrated for p.R39Q and p.R367Q in supplemental Figures 2 and 3).31 The third base mismatch primers outperformed those with second base mismatches (supplemental Figure 2).30 From the former primer set, those with the best ASQ-PCR performance were selected, evaluated for the degree of unspecific amplification (supplemental Table 4), and optimized for annealing temperature (supplemental Figure 4). For quantification of NT5C2 mutations, the ratio of mutation-positive cells to wild-type cells in a patient sample was calculated using standard curve equations. For sensitive screening of low-frequency NT5C2 p.R39Q and p.R367Q mutations, a background cutoff cycle threshold (CT) was defined following the minimal residual disease detection guidelines.31 The median limit of detection was 3E-05 for the p.R39Q and 4E-05 for the p.R367Q ASQ-PCR assay. Experimental details are specified in supplemental Methods. The ASQ-PCR values for p.R39Q and p.367Q correlated well with the variant allele frequency determined by NGS in 16 relapses (r2 = 0.846; supplemental Figure 5).
Descriptive statistics and survival analyses
All statistical calculations were performed using the SPSS software version 25.0.0 (SPSS, Chicago, IL). The level of significance was set to 0.05. Relationships between NT5C2 mutations and clinical and genetic parameters were assessed by χ2 or Fisher’s exact test for categorical variables and by Mann-Whitney U or Kruskal-Wallis test for continuous variables. Data on genetic alterations in relapses of the present study were largely available from our previous work with detailed information given in the supplemental Methods. The distribution of recurrent genetic alterations in relapses with clonal and with subclonal NT5C2 mutations was visualized using the cBioPortal OncoPrinter.32 For outcome analyses, patients taken off the ALL-REZ BFM 2002 trial due to protocol violation were excluded (12 of 455 patients, including patient ID 444 with clonal NT5C2 mutation). EFS and overall survival (OS) were calculated by Kaplan-Meier analysis and differences were assessed with the log-rank test. EFS time was defined as the time from relapse to subsequent event (second relapse, secondary malignancy, or death in complete remission) or to the time of data analysis for patients in continuous complete remission. In case of death during induction treatment or nonresponse to chemotherapy, the time to event was set to zero. OS time was defined as the time from relapse to death or to the time of data analysis. The cumulative incidence of subsequent relapse (CIR) and the cumulative incidence of therapy-related death (CID) were assessed using the Kalbfleisch-Prentice method and Gray statistics and R statistical software (version 3.6.1). All EFS, OS, CIR, and CID rates were quoted at 10 years. Cox regression modeling was performed for EFS and OS by a backward-selection stepwise modeling process. Differences in covariate effects were assessed by the Wald test. The proportional hazards assumption was verified for all variables by log-minus-log survival plots and a sensitivity analysis was performed (supplemental Methods).
Results
Frequency and distribution of NT5C2 mutations in relapsed BCP-ALL
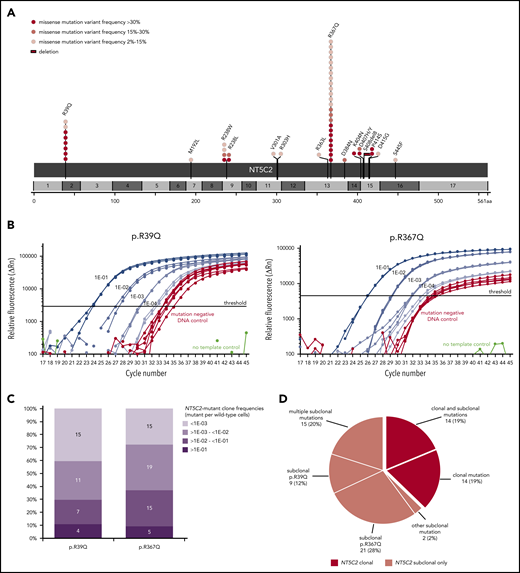
For comprehensive identification of NT5C2 mutations, we first applied deep amplicon-based NGS covering all NT5C2 exons to a subset of 228 relapses that were representative for the total ALL-REZ BFM 2002 cohort of isolated and combined bone marrow relapses of BCP-ALL (n = 552) with respect to the most relevant clinical factors (supplemental Table 1). With a variant allele frequency threshold of 2%, NGS identified 32 mutations in 27 (12%) of 228 relapses that could be validated by Sanger sequencing and/or sensitive ASQ-PCR (supplemental Tables 5 and Table 6). Consistent with published data,21,22 >90% (29/32) of NGS-detected mutations mapped to the NT5C2 hotspot exons 2, 9, 13, 15, and 16. Sequencing of these exons in an independent subset of 227 relapses using the cost-efficient method of Sanger identified additional 19 mutations in 19 (8%) of 227 relapses (supplemental Table 5). Collectively, NGS and Sanger sequencing detected 51 NT5C2 mutations in 46 (10%) of 455 relapses (Figure 1A). The most prevalent mutant allele was p.R367Q (c.1100G>A) followed by p.R39Q (c.116G>A), which together accounted for 63% of all NT5C2 mutations. Other sites of recurrent mutation were codons 238 and 404 to 415 (Figure 1A). In total, we identified 16 different mutant alleles, including 15 missense mutations and 1 in-frame deletion (p.S408del8).
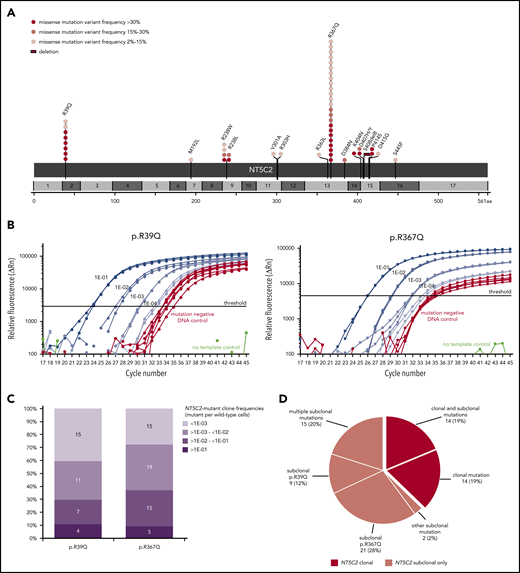
NT5C2 mutations in relapsed pediatric BCP-ALL. (A) Schematic representation of the NT5C2 protein/gene. Mutations identified by Sanger sequencing and/or targeted next generation in relapses of 455 patients with BCP-ALL are described at the protein level. Multiple circles in the same amino acid position account for multiple patients with the same variant. NT5C2 exons are indicated by alternating light and dark gray boxes. Variant frequency refers to variant allele frequency for NGS-detected mutations and allelic peak ratio for Sanger-detected mutations. (B) Performance of the ASQ-PCR assays developed for sensitive detection of NT5C2 mutations p.R39Q and p.R367Q. The graphs represent exemplary amplification plots of standard dilution series of NT5C2 mutation-positive patient samples (blue) as well as of a mutation-negative DNA control (red). The distance between the 1E-03 dilution and the mutation-negative DNA control is larger than 3 CT values for both assays as required by the guidelines for minimal residual disease detection in leukemia.31 (C) Range of NT5C2-mutant clone frequencies determined by ASQ-PCR for all p.R39Q and p.R367Q mutations. For this graphic, NT5C2-mutant clone frequencies were categorized into different quantitative levels in log10 steps. The bar graphs represent the percentage of subclonal NT5C2 p.R39Q and p.R367Q mutations with a given quantitative level. The absolute number of mutations is displayed within the bar graphs. (D) Classification of relapsed patients with NT5C2 mutations after compilation of sequencing and ASQ-PCR data into 2 main groups: patients with clonal NT5C2 mutation (dark-red) and patients with subclonal NT5C2 mutation(s) only (light red). Approximately one-third of relapses harbored >1 NT5C2 mutation.
NT5C2 mutations in relapsed pediatric BCP-ALL. (A) Schematic representation of the NT5C2 protein/gene. Mutations identified by Sanger sequencing and/or targeted next generation in relapses of 455 patients with BCP-ALL are described at the protein level. Multiple circles in the same amino acid position account for multiple patients with the same variant. NT5C2 exons are indicated by alternating light and dark gray boxes. Variant frequency refers to variant allele frequency for NGS-detected mutations and allelic peak ratio for Sanger-detected mutations. (B) Performance of the ASQ-PCR assays developed for sensitive detection of NT5C2 mutations p.R39Q and p.R367Q. The graphs represent exemplary amplification plots of standard dilution series of NT5C2 mutation-positive patient samples (blue) as well as of a mutation-negative DNA control (red). The distance between the 1E-03 dilution and the mutation-negative DNA control is larger than 3 CT values for both assays as required by the guidelines for minimal residual disease detection in leukemia.31 (C) Range of NT5C2-mutant clone frequencies determined by ASQ-PCR for all p.R39Q and p.R367Q mutations. For this graphic, NT5C2-mutant clone frequencies were categorized into different quantitative levels in log10 steps. The bar graphs represent the percentage of subclonal NT5C2 p.R39Q and p.R367Q mutations with a given quantitative level. The absolute number of mutations is displayed within the bar graphs. (D) Classification of relapsed patients with NT5C2 mutations after compilation of sequencing and ASQ-PCR data into 2 main groups: patients with clonal NT5C2 mutation (dark-red) and patients with subclonal NT5C2 mutation(s) only (light red). Approximately one-third of relapses harbored >1 NT5C2 mutation.
Interestingly, a substantial proportion of NT5C2 mutations appeared subclonal with low variant allele frequencies (NGS) or low allelic peak ratios (Sanger) of <15% (23 [45%] of 51; Figure 1A; supplemental Figure 1). To investigate subclonal NT5C2 mutations in more detail, we used the best-performing ASQ-PCR assays targeting the 2 most frequent mutant alleles in our cohort (p.R39Q and p.R367Q; Figure 1B; supplemental Figures 2-4; supplemental Tables 3 and 4) and applied them to all 455 relapses for highly sensitive detection of low-frequency mutations. ASQ-PCR identified 29 relapses that had been wild-type by NGS and Sanger sequencing to harbor 39 low-frequency p.R39Q and/or p.R367Q mutations (supplemental Table 5). Besides, ASQ-PCR identified 18 relapses that had shown NT5C2 mutations by NGS or Sanger sequencing before to harbor 20 additional low-frequency p.R39Q and/or p.R367Q mutations below the NGS/Sanger sequencing detection limit (supplemental Table 5). Quantification of all p.R39Q and p.R367Q mutant subclones (<15% variant allele frequency or allelic peak ratio) by ASQ-PCR revealed a broad range of quantitative levels from >1E-01 up to the detection limits of the assays (Figure 1C). The majority of NT5C2-mutant subclones was present below the 1E-02 level (p.R39Q: 26 [70%] of 37; p.R367Q: 34 [63%] of 54; Figure 1C; supplemental Table 5). Compiling sequencing and ASQ-PCR data, we found a total of 110 NT5C2 mutations in 75 (16.5%) of 455 BCP-ALL relapses. More than one-third of NT5C2-mutated relapses (29 [39%] of 75) harbored double (n = 26) or multiple NT5C2 mutations (n = 3) including co-occurring subclonal (n = 15) as well as co-occurring subclonal and clonal mutations (n = 14) (Figure 1D; supplemental Table 5). Of note, 63% of NT5C2-mutated relapses (47 of 75) harbored subclonal NT5C2 mutations only (Figure 1D).
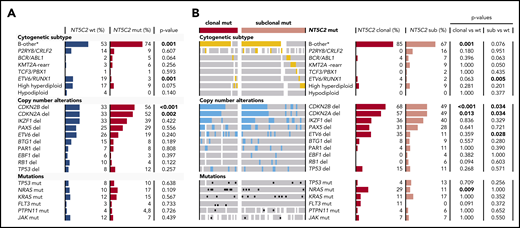
We detected NT5C2 mutations in all established cytogenetic subtypes of ALL except the hypodiploid group (Figure 2A; supplemental Table 7). However, the favorable prognostic ETV6-RUNX1 fusion gene11,26 was rare in relapses with NT5C2 mutations compared with those without (3% vs 19%, P = .001). In contrast, the B-other-ALL subtype, which lacks established cytogenetic changes,33 occurred more frequently in relapses with NT5C2 mutations than in those without (74% vs 53%, P = .001). Deletions of CDKN2A and CDKN2B were more often present in relapses with NT5C2 mutations than in those without (52% vs 33%, P = .002 and 56% vs 33%, P < .001, respectively). Differentiating between relapses with clonal and subclonal NT5C2 mutations (Figure 2B; supplemental Table 7), we found an ∼3 times increased NRAS mutation rate in relapses with clonal, but not with subclonal, NT5C2 mutations as compared with relapses with wild-type NT5C2 (29% vs 10%, P = .009 and 10% vs 11%, P = 1, respectively). However, the presence of subclonal NRAS/KRAS mutations, which occur frequently in ALL,34 was not associated with the presence of clonal or subclonal NT5C2 mutations (supplementary Figure 6). The enrichments of the B-other ALL subtype and of CDKN2A/2B deletions in relapses with NT5C2 mutations were strongly pronounced in the subgroup with clonal NT5C2 mutations with 58% and 68% of relapses harboring a CDKN2A and CDKN2B deletion, respectively, and 85% belonging to the B-other ALL subtype (Figure 2B; supplemental Table 7).

Association of NT5C2 mutations with recurrent genetic alterations in relapsed BCP-ALL. (A) Distribution of recurrent genetic alterations in relapses with wild-type NT5C2 in comparison with relapses with NT5C2 mutation. Bars represent the percentage of relapses with a given genetic alteration within each category. P values below the .05 level of significance are depicted in bold. (B) Distribution of recurrent genetic alterations in relapses with clonal and with subclonal NT5C2 mutations only in comparison with relapses with wild-type NT5C2. Colored bars (yellow and blue) and black dots highlight the presence of a given genetic alterations in a relapse sample, whereas white bars indicate cases with no data. del, deletion; mut, mutation; rearr, rearrangement; sub, subclonal NT5C2 mutation; wt, wild-type. B-other* represents BCP-ALL relapses lacking the established cytogenetic abnormalities listed below.
Association of NT5C2 mutations with recurrent genetic alterations in relapsed BCP-ALL. (A) Distribution of recurrent genetic alterations in relapses with wild-type NT5C2 in comparison with relapses with NT5C2 mutation. Bars represent the percentage of relapses with a given genetic alteration within each category. P values below the .05 level of significance are depicted in bold. (B) Distribution of recurrent genetic alterations in relapses with clonal and with subclonal NT5C2 mutations only in comparison with relapses with wild-type NT5C2. Colored bars (yellow and blue) and black dots highlight the presence of a given genetic alterations in a relapse sample, whereas white bars indicate cases with no data. del, deletion; mut, mutation; rearr, rearrangement; sub, subclonal NT5C2 mutation; wt, wild-type. B-other* represents BCP-ALL relapses lacking the established cytogenetic abnormalities listed below.
Clinical effects of NT5C2 mutations in relapsed BCP-ALL
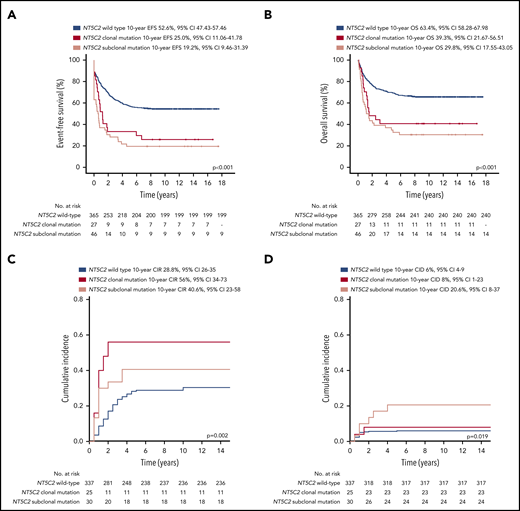
Patients with relapses with either clonal or subclonal NT5C2 mutations showed a significantly reduced median time between initial diagnosis and first relapse compared with those without (1.6 years and 1.7 years vs 3.1 years, P < .001; Table 1). Accordingly, the proportion of relapses that occurred very early or early after initial diagnosis of ALL was ∼3 times increased in the clonal (39% and 53%) as well as in the subclonal NT5C2 mutation group (36% and 60%) compared with relapses with wild-type NT5C2 (12% and 18%, P < .001; Table 1). Similarly, the proportion of relapses that occurred while the patients were still being treated for primary ALL was strongly increased to 89% and 75% in relapses harboring clonal and subclonal NT5C2 mutations, respectively, compared with 19% in relapses with wild-type NT5C2 (P < .001; Table 1). Consequently, 75% and 77% of patients with relapses harboring either clonal or subclonal NT5C2 mutations, respectively, were stratified into the high-risk S3/S4 treatment arm of the ALL-REZ BFM 2002 protocol compared with only 24% of patients with wild-type NT5C2 relapses (P < .001; Table 1). Despite high-risk relapse treatment, EFS after relapse was significantly reduced in patients with relapses with either clonal or subclonal NT5C2 mutations compared with those without in Kaplan-Meier analysis (25% [95% confidence interval (CI), 11.06-41.78] and 19.2% [95% CI, 9.46-31.39], respectively, vs 52.6% [95% CI, 47.43-57.46], P < .001; Figure 3A). The same association was found for OS (Figure 3B). The cumulative incidence of relapse was highest in patients with relapses with clonal NT5C2 mutations (clonal, 56% [95% CI, 34-73]; subclonal, 41% [95% CI 23-58]; and wild-type, 28.8% [95% CI 26-35]; P = .002; Figure 3C), consistent with a second relapse being the most frequent second event in this patient group (Table 2). The cumulative incidence of death was slightly increased in patients with relapses with subclonal NT5C2 mutations (subclonal, 21% [95% CI, 8-37]; clonal, 8% [95% CI, 1-23]; and wild-type, 6% [95% CI 4-9]; P = .019; Figure 3D). Remarkably, in patients with relapses with subclonal, but not clonal, NT5C2 mutations, the rate of nonresponse to relapse treatment was increased to 32% as compared with 12% or 9% in patients with relapses with clonal NT5C2 mutations or with wild-type (P = .001; Table 2). Moreover, of those relapsed patients with subclonal NT5C2 mutations who responded to relapse treatment, 76% presented with high minimal residual disease levels at the end of relapse induction treatment compared with 53% and 50% of patients with relapses with clonal NT5C2 mutations and with wild-type NT5C2, respectively (P = .030; Table 2). Multivariable Cox regression analysis for EFS, including clonal and subclonal NT5C2 mutation in addition to sex and the established risk factors time between initial diagnosis and first relapse, site of relapse, and TP53 mutation/deletion as well as allo-HSCT as a time-dependent covariate, identified subclonal NT5C2 mutation, but not clonal NT5C2 mutation, as an independent risk factor for the occurrence of a second event after relapse next to the time to relapse, allo-HSCT, and TP53 mutation/deletion (hazard ratio [HR], 1.89; 95% CI, 1.28-2.69; P = .001; Table 3). For OS, subclonal NT5C2 mutations were not significantly predictive in multivariable analysis (HR, 1.4; 95% CI, 0.93-2.16; P = .103; supplemental Table 9). We performed a sensitivity analysis examining the influence of allo-HSCT and of different covariates (eg, time between initial diagnosis and first relapse as continuous versus categorical variable) on the Cox regression model and found that the results were robust (supplemental Tables 8 and 9).
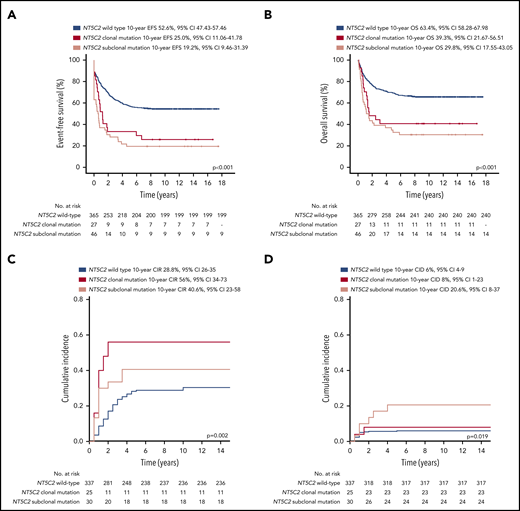
Survival outcomes of patients after first relapse of BCP-ALL by NT5C2 mutation status. (A) Event-free survival (EFS). (B) Overall survival (OS). (C) Competing risk analysis of cumulative incidence of relapse (CIR) at 10 years. (D) Competing risk analysis of cumulative incidence of treatment related death (CID) at 10 years. cens, censored.
Survival outcomes of patients after first relapse of BCP-ALL by NT5C2 mutation status. (A) Event-free survival (EFS). (B) Overall survival (OS). (C) Competing risk analysis of cumulative incidence of relapse (CIR) at 10 years. (D) Competing risk analysis of cumulative incidence of treatment related death (CID) at 10 years. cens, censored.
Evolutionary trajectories of NT5C2 mutations during relapse progression
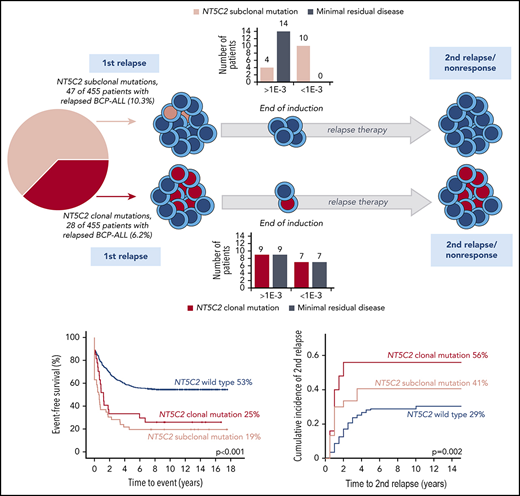
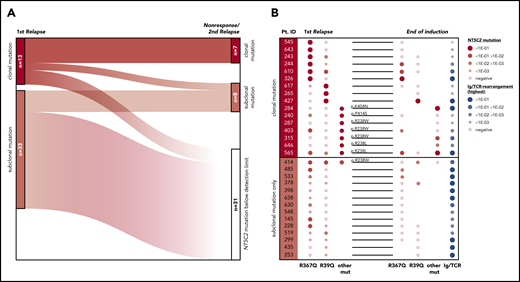
To investigate the development of NT5C2-mutant leukemic clones during and after relapse therapy, we examined follow-up samples of the patients for NT5C2 mutations. First, we determined whether NT5C2 mutations were retained in samples taken at the time of nonresponse to treatment or second relapse from 12 patients with clonal and 22 patients with only subclonal NT5C2 mutations at first relapse (supplemental Figure 7). By ASQ-PCR, we found that 54% (7 of 13) of the clonal NT5C2 mutations persisted as clonal mutations at the time of nonresponse or second relapse, whereas 46% (6 of 13) were reduced to a subclonal mutation or became undetectable (Figure 4A; supplemental Figure 7). By contrast, only 18% (6 of 33) of all subclonal mutations (including those co-occurring with clonal NT5C2 mutations; see supplemental Figure 7) persisted as subclonal mutations, whereas the great majority of 82% (27 of 33) fell below the detection limit at the time of nonresponse or second relapse (Figure 4A; supplemental Figure 7). Sanger sequencing of all NT5C2 hotspot exons in nonresponse or second relapse samples of patients who lost the NT5C2 mutations from first relapse showed that these samples did not acquire new clonal NT5C2 mutations, consistent with the outgrowth of NT5C2 wild-type cells at the time of nonresponse or second relapse. To gain a deeper insight into the response of NT5C2-mutant clones to relapse therapy, we examined samples taken at the end of relapse induction treatment. At this time point (∼4 weeks after begin of relapse therapy), minimal residual disease levels are prognostic for patient outcome.7 By ASQ-PCR, we quantified the end induction levels of p.R39Q, p.R238W, p.R238L, p.R367Q, p.K404N, and p.P414S in a subset of 16 patients with clonal and of 14 patients with subclonal NT5C2 mutations only and compared them to minimal residual disease levels (assessed by immunoglobulin and T-cell receptor gene rearrangement quantification in routine diagnostics) as a measure of the overall leukemic burden in the patients (Figure 4B). Levels of clonal NT5C2 mutations at the end of induction treatment were heterogeneous, with 7 (44%) of 16 lying below and 9 (56%) of 16 above the prognostic minimal residual disease cutoff value of 1E-03,7 corresponding to the minimal residual disease levels of the patients (Figure 4B). By contrast, in patients with subclonal NT5C2 mutations at first relapse only, minimal residual disease and NT5C2 mutation levels diverged at the end of induction treatment. Whereas NT5C2 mutation levels were frequently reduced to below the detection limit (71%, 10 of 14 cases), minimal residual disease levels remained high (>1E-03) in all cases (Figure 4B), further supporting the view that NT5C2 wild-type, but not NT5C2-mutant leukemic, cells persist and outgrow in these patients.
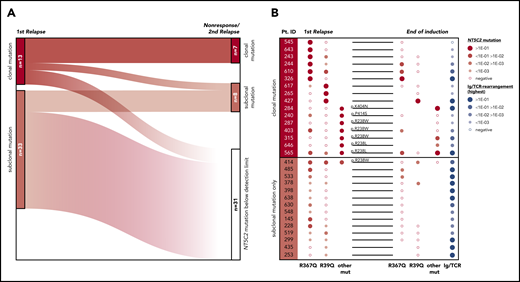
Evolutionary trajectories of NT5C2 mutations after first relapse. (A) The Sankey diagram illustrates the fate of 46 NT5C2 mutations from 34 first relapses in follow-up samples taken at the time of nonresponse to treatment or second relapse of the patients. All 46 mutations were tracked sensitively by p.R39Q, p.R238W, p.R367Q, p.K404N, or p.P414S ASQ-PCR. (B) Quantitative levels of NT5C2 mutations (red dots) at first relapse and at the end of relapse induction treatment in 16 patients with clonal and in 14 patients with subclonal mutations only. All NT5C2 mutations were tracked sensitively by p.R39Q, p.R238W, p.R238L, p.R367Q, p.K404N, or p.P414S ASQ-PCR. Minimal residual disease levels (blue dots) were assessed by immunoglobulin/ T-cell receptor (Ig/TCR) gene rearrangement quantification and are given as a measure of the overall leukemic burden in the patients at the end of induction treatment. Pt. ID, patient identifier.
Evolutionary trajectories of NT5C2 mutations after first relapse. (A) The Sankey diagram illustrates the fate of 46 NT5C2 mutations from 34 first relapses in follow-up samples taken at the time of nonresponse to treatment or second relapse of the patients. All 46 mutations were tracked sensitively by p.R39Q, p.R238W, p.R367Q, p.K404N, or p.P414S ASQ-PCR. (B) Quantitative levels of NT5C2 mutations (red dots) at first relapse and at the end of relapse induction treatment in 16 patients with clonal and in 14 patients with subclonal mutations only. All NT5C2 mutations were tracked sensitively by p.R39Q, p.R238W, p.R238L, p.R367Q, p.K404N, or p.P414S ASQ-PCR. Minimal residual disease levels (blue dots) were assessed by immunoglobulin/ T-cell receptor (Ig/TCR) gene rearrangement quantification and are given as a measure of the overall leukemic burden in the patients at the end of induction treatment. Pt. ID, patient identifier.
Discussion
The present study contributes a comprehensive analysis of the clinical effects of NT5C2 mutations in pediatric patients with relapsed BCP-ALL. The study cohort includes 70% of the total ALL-REZ BFM 2002 trial cohort, has a median long-term follow-up of nearly 12 years, and no bias for clinical subgroups except the 15% of patients with extramedullary relapses that were excluded due to scarce sample availability. Our study confirms p.R367Q as most frequent NT5C2 mutation in relapsed BCP-ALL.21,22 Thirteen of the 16 different mutant alleles identified in the ALL-REZ BFM 2002 cohort have been described previously as gain-of-function alleles in relapsed ALL and/or mapped to NT5C2 areas that represent mutation hotspots with a role in nucleotidase activation (summarized in supplemental Table 10).14-19,21,22,27 Only 3 mutant alleles have not been further annotated so far (V301A, R303H, and D384N).
Two-thirds of NT5C2 mutations in our study were subclonal, and only one-third were clonal. This finding validates results from 2 earlier studies that described subclonal NT5C2 mutations in 67% of 20 patients with relapsed BCP-ALL16 and 71% of 67 patients with relapsed T-ALL.18 Moreover, more than one-third of relapses harbored multiple NT5C2 mutations, consistent with an earlier study by Ma et al, who modeled these mutations to occur in distinct leukemic clones based on deep sequencing data.16 The subclonal nature of NT5C2 mutations in relapsed ALL may be attributed to a loss-of-fitness phenotype of NT5C2-mutant leukemic cells that was identified in a NOTCH1-induced T-ALL mouse model.19 In this model, mutant NT5C2 caused imbalances in the intracellular nucleotide pool, impaired leukemia cell growth, and leukemia-initiating cell activity but provided a selective growth advantage under 6-mercaptopurine treatment.19 Likewise, NT5C2 mutations may suppress leukemia cell growth in patients and cause NT5C2-mutant cells to remain at subclonal levels. However, different from the model, NT5C2 mutation may be insufficient to overcome the selective pressure of 6-mercaptopurine and methotrexate based ALL maintenance therapy in patients,24 and distinct cooperative events may be required to allow NT5C2-mutant cells to escape from negative selection during relapse formation. The pronounced enrichment of NRAS mutations, CDKN2A/B deletions and the B-other ALL subtype in relapses with clonal, but not subclonal, NT5C2 mutations in our study supports this hypothesis.
Clinically, the results of our study firmly link NT5C2 mutations with early disease recurrence and with on-treatment relapse in pediatric BCP-ALL.14,15,18 Beyond that, they first link NT5C2 mutations with poor EFS and OS in relapsed BCP-ALL. The only previous analysis of the impact of NT5C2 mutations on survival of patients with relapsed leukemia was one report that found no enrichment of NT5C2 mutations in T-ALL relapses from patients who suffered a second event after relapse treatment, concluding that NT5C2 mutation had no prognostic impact in the 67 patients analyzed.18 However, T-lineage immunophenotype itself is strongly associated with inferior survival rates after relapse compared with BCP-ALL,3-5 which may mask an adverse prognostic effect of NT5C2 mutation in relapsed T-ALL. Intriguingly, subclonal, but not clonal, NT5C2 mutations were (1) independently associated with inferior EFS in multivariable analysis and (2) had an increased rate of nonresponse to relapse treatment in our study. Collectively, this suggests that patients with relapses harboring subclonal NT5C2 mutations represent a previously unidentified clinical subgroup with high risk of relapse therapy failure. However, subclonal NT5C2 mutations never developed into clonal mutations during or after relapse therapy but frequently became undetectable already after relapse induction treatment. Instead, NT5C2 wild-type leukemic cells persisted in patients with relapses harboring subclonal NT5C2 mutations and formed the nonresponding leukemia or the second relapse. These findings support a more complex situation leading to treatment failure after relapse than subclonal NT5C2 mutations driving the persistence and expansion of resistant leukemic clones.35
In view of the rapid eradication of NT5C2-mutant subclones by standard relapse polychemotherapy, incorporating targeted treatment of mutant NT5C2 or of collaterally affected pathways12,19 into the treatment of relapses harboring subclonal NT5C2 mutations is not a reasonable therapeutic strategy to improve the outcome of this patient subgroup. Nevertheless, for relapsed patients with clonal NT5C2 mutations in whom standard relapse therapy fails, targeting mutant NT5C2 may be considered in a third-line personalized treatment approach,36 potentially in combination with RAS effector pathway inhibition37 in case of a co-occurring RAS pathway mutation. Whether earlier targeting of mutant NT5C2 already during maintenance therapy of primary ALL may be effective in reducing the occurrence of NT5C2-mutated relapses12 remains to be elucidated. The predominantly subclonal nature of NT5C2 mutations in relapsed ALL supports the view that their role in relapse formation and resistance to chemotherapy is complex and goes beyond conferring 6-mercaptopurine resistance to the leukemic cells in which they occur. We recommend that systematic prospective identification of NT5C2 mutations in ALL patients be integrated into trials in planning to support an improved understanding of the aggressive relapses harboring NT5C2 mutations, which is essential for the design and support of future treatment approaches for patients with ALL.
All primers and data on the optimization of ASQ-PCR may be found in a data supplement available with the online version of this article. For original data, please contact r.kirschner@charite.de.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by German Childhood Cancer Foundation grants DKS-2015.07 (R.K.-S. and C.E.) and DKS-2007.02 (A.v.S.), a Rachel-Hirsch fellowship of the Charité–Universitätsmedizin Berlin (R.K.-S.), and a fellowship of the Sonnenfeld-Stiftung Berlin (M.J.P.). J.-W.L. is a predoctoral fellow of the New Jersey Commission on Cancer Research (DFS18PPC017). H.K. is supported by the National Institutes of Health (NIH) National Cancer Institute (grant R01CA233662) and the V Foundation (grant T2019-012), and also acknowledges support from Rutgers Cancer Institute of New Jersey Bioinformatics Shared Resource (P30CA072720-5917) as well as Rutgers Office of Advanced Research Computing (NIH Office of the Director grant 1S10OD012346-01A1).
Authorship
Contribution: M.J.B., J.H., S.G.-K., A.v.S., and R.K.-S. designed the study and performed research; M.J.B., J.-W.L., H.K., and R.K.-S. performed data analysis; C.E. and A.v.S. provided clinical data; A.A.F., A.v.S., K.A., C.E., and R.K.-S. supervised the study; M.J.B., K.A., K.H., and R.K.-S. wrote the manuscript; M.J.B. and R.K.-S. had the final responsibility to submit for publication; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Renate Kirschner-Schwabe, Department of Pediatric Oncology and Hematology, Charité–Universitätsmedizin Berlin, Campus Virchow, Augustenburger Platz 1, 13353 Berlin, Germany; e-mail: r.kirschner@charite.de.