

Key Points
Transplant survival for MHC class II deficiency has improved significantly in the modern era of HCT.
Long-term health outcomes after HCT for MHC class II expression deficiency are good.

Abstract
MHC class II deficiency is a rare, but life-threatening, primary combined immunodeficiency. Hematopoietic cell transplantation (HCT) remains the only curative treatment for this condition, but transplant survival in the previously published result was poor. We analyzed the outcome of 25 patients with MHC class II deficiency undergoing first HCT at Great North Children's Hospital between 1995 and 2018. Median age at diagnosis was 6.5 months (birth to 7.5 years). Median age at transplant was 21.4 months (0.1-7.8 years). Donors were matched family donors (MFDs; n = 6), unrelated donors (UDs; n = 12), and haploidentical donors (HIDs; n = 7). Peripheral blood stem cells were the stem cell source in 68% of patients. Conditioning was treosulfanbased in 84% of patients; 84% received alemtuzumab (n = 14) or anti-thymocyte globulin (n = 8) as serotherapy. With a 2.9-year median follow-up, OS improved from 33% (46-68%) for HCT before 2008 (n = 6) to 94% (66-99%) for HCT after 2008 (n = 19; P = .003). For HCT after 2008, OS according to donor was 100% for MFDs and UDs and 85% for HIDs (P = .40). None had grade III-IV acute or chronic graft-versus-host disease. Latest median donor myeloid and lymphocyte chimerism were 100% (range, 0-100) and 100% (range, 64-100), respectively. Latest CD4+ T-lymphocyte number was significantly lower in transplant survivors (n = 14) compared with posttransplant disease controls (P = .01). All survivors were off immunoglobulin replacement and had protective vaccine responses to tetanus and Haemophilus influenzae. None had any significant infection or autoimmunity. Changing transplant strategy in Great North Children's Hospital has significantly improved outcomes for MHC class II deficiency.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 975.
Disclosures
Associate Editor Catherine M. Bollard served as an advisor or consultant for Cellectis, NovImmune SA, and Roche and owns stocks, stock options, or bonds from Cabaletta Bio, Mana Therapeutics, NexImmune Inc., and Torque Therapeutics. CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC and the authors declare no relevant financial relationships.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe transplant survival of children with major histocompatibility complex (MHC) class II deficiency transplanted at a single national center, according to a retrospective study
Determine long-term disease outcomes of children with MHC class II deficiency transplanted at a single national center, according to a retrospective study
Identify clinical implications of transplant survival and long-term disease outcomes of children with MHC class II deficiency transplanted at a single national center, according to a retrospective study
Release date: March 19, 2020; Expiration date: March 19, 2021
Introduction
Major histocompatibility complex (MHC) class II deficiency is a rare autosomal recessive combined immunodeficiency.1 MHC class II genes are located on chromosome 6, and expression is largely restricted to activated T lymphocytes, thymic epithelial cells, and antigen-presenting cells (dendritic cells, macrophages, and B lymphocytes). In patients with MHC class II deficiency, the MHC locus itself is intact but transcriptionally silent because of loss-of-function mutations in 1 of 4 genes encoding the key regulatory factors: CIITA (class II transactivator), RFX5 (regulatory factor 5), RFXAP (RFX-associated protein), and RFXANK (RFX-associated ankyrin-containing protein). MHC class II molecules are pivotal for the adaptive immune system and guide the development and function of CD4+ T lymphocytes. The immunologic hallmark of the disease is the absence of constitutive and inducible expression of MHC class II molecules on all cell types, which leads to impaired antigen presentation by HLA-DR, HLA-DQ, and HLA-DP molecules on antigen-presenting cells.2 The lack of MHC class II expression on thymic epithelium leads to delayed and incomplete maturation of the CD4+ T-lymphocyte population. Overall, MHC class II deficiency leads to combined immunodeficiency with defective CD4+ T-lymphocyte maturation and activation and a lack of T-helper lymphocyte-dependent antibody production by B lymphocytes, resulting in significant susceptibility to severe infections.3
Hematopoietic cell transplantation (HCT) is the only curative therapy for children with MHC class II deficiency. The natural history of nontransplanted patients is dismal, and the main cause of death is overwhelming viral infection.4 Very few children survive into adulthood.5 HCT for MHC class II deficiency is challenging because many children have significant comorbidities at the time of HCT, increasing their susceptibility to regimen-related toxicities, serious infections, graft rejection, and graft-versus-host disease (GvHD). Historically, the use of HCT has been limited because of the high risk of transplant-related morbidity and mortality. Reported transplant survival was poor compared with that seen in children with classical severe combined immunodeficiency (SCID), with a survival rate ≤ 50%.3,6-9 In light of the significant improvements in transplant care for children with primary immunodeficiency (PID) over time, the present retrospective study aimed to examine transplant survival and long-term disease outcomes of children with MHC class II deficiency transplanted at a single national center.
Methods
From January of 1995 to December of 2018, a total of 25 children with MHC class II deficiency underwent first HCT at Great North Children’s hospital, Newcastle upon Tyne, United Kingdom. The diagnosis of MHC class II deficiency was confirmed by the absence of DR expression on monocytes, B lymphocytes, and T lymphocytes. The clinical and laboratory data were retrieved from the transplantation database, patients’ medical files, and laboratory records. Long-term follow-up outcomes from overseas patients were collected from the respective referral hospitals. Written informed consent was obtained from the parents or legal guardians of the patients, as per the institutional practice for HCT.
Donor selection, stem cell source, conditioning regimen, and GvHD prophylaxis
The donor selection hierarchy was as follows: (1) matched family donor (MFD), (2) matched unrelated donor (MUD), (3) single-antigen mismatched unrelated donor (MMUD), (4) haploidentical donor (HID). For unrelated donor (UD) grafts, donor-recipient matching was by allele-level typing at HLA-A, HLA-B, HLA-C, HLA-DQ and HLA-DR. Prior to 2009, the conditioning regimen was busulfan or treosulfan in combination with cyclophosphamide with or without serotherapy. From 2009, the conditioning regimen was switched to a myeloablative reduced toxicity conditioning (RTC) regimen using fludarabine, treosulfan, and alemtuzumab for MFDs and MUDs, whereas fludarabine, treosulfan, thiotepa, anti-thymocyte globulin (ATG; Grafalon), and rituximab were used for TCRαβ/CD19-depleted parental grafts. Marrow was the conventional preferred stem cell source, but peripheral blood stem cells (PBSCs) were preferred for patients who received RTC with matched donors or TCRαβ/CD19-depleted parental grafts. Before 2008, GvHD prophylaxis regimens varied but consisted primarily of cyclosporine A (CSA) alone or in combination with methotrexate (MTX). From 2008, GvHD prophylaxis shifted almost exclusively to CSA and mycophenolate mofetil (MMF). From 2017, the recipients of TCRαβ/CD19-depleted parental grafts did not receive any posttransplant GvHD prophylaxis.
Supportive care
Prior to transplant, bronchoalveolar lavage samples from all patients were screened by polymerase chain reaction for viremia, gut viruses, and respiratory viruses. Surveillance for cytomegalovirus (CMV), adenovirus, Epstein-Barr virus (EBV), human herpesvirus 6 (HHV6) viremia, and respiratory and gut viruses was performed weekly since 2000. All patients received antimicrobial prophylaxis against fungi, Pneumocystis jirovecii pneumonia (PCP), and human herpesvirus reactivation. All patients received immunoglobulin (Ig) replacement until normal IgM was made. Donor hematopoietic chimerism was monitored by molecular techniques.
Definition and end points
The main outcomes of interest were overall survival (OS) and event-free survival (EFS). OS was defined as survival from first HCT to last follow-up or death. An event was defined as death, graft failure, or second procedure for slipping chimerism. Other end points assessed were as follows: time to neutrophil recovery (first day of achieving a neutrophil count ≥0.5 × 109/L for 3 consecutive days); time to platelet recovery (platelets >20 × 109/L without transfusions for 7 days); transplant-related complications, as defined and graded according to existing institutional guidelines at the time of HSCT, including infections, hepatic veno-occlusive disease, and GvHD; degree of donor hematopoietic chimerism at the most recent assessment; and immune reconstitution.
Statistical analysis
Quantitative variables are described using median and range, whereas categorical variables are reported with counts and percentages. The χ2 test was used to compare categorical variables. Probabilities of OS and EFS were calculated using the Kaplan-Meier estimate. The log-rank test was used to compare predictors on OS. Variables analyzed included age at transplant, year of transplant (1995-2008 vs 2009-2018), donor type, stem cell source, and stem cell dose. For immune reconstitution kinetics for the first 12 months and the latest lymphocyte subset results, a nested matched case-control study was performed in which each MHC class II–deficient patient was matched with 2 disease controls for the following variables: age (difference <2 years at transplant for immune reconstitution kinetics and at last review for latest lymphocyte subset), donor type, stem cell source, conditioning regimen, serotherapy, GvHD prophylaxis, and CMV reactivation. Disease controls (n = 36) were patients who were transplanted for alternative PID diagnoses: SCID (n = 10), CD40 ligand deficiency (n = 2), Wiskott-Aldrich syndrome (n = 7), chronic granulomatous disease (n = 13) and other PIDs (n = 4; 1 natural killer [NK] deficiency, 1 inducible costimulator deficiency, 1 hemophagocytic lymphohistiocytosis, 1 combined immunodeficiency). Lymphocyte subset at last review was available for 14 of 19 survivors (1 was on active treatment for GvHD, 1 was <1 year post-HCT at the time of analysis, and 3 overseas patients were not able to be contacted). Multilevel mixed-effects modeling was performed for the longitudinal analysis of CD3+, CD4+, CD8+, CD19+, and NK cells, as well as HLA-DR. The Wilcoxon rank-sum test was used to compare latest lymphocyte subsets between patients and disease controls, as above. All quoted P values are 2-sided, with a level of significance of .05. Statistical analyses were performed using STATA 14.2.
Results
Patient characteristics
Patient clinical and immunological features at presentation are shown in Tables 1 and 2. The median age at diagnosis was 6.5 months (range, at birth to 7.5 years). Immunological evaluation performed at referral showed that 14 (56%) had lymphocytopenia, 10 (40%) had normal lymphocyte counts, and 1 had lymphocytosis. CD4+ lymphocytopenia was present in 19 patients (76%), and 6 (24%) had a normal CD4+ lymphocyte count. Six (24%) had CD19 lymphocytopenia. All but 4 patients who were diagnosed at birth had multiple infections. Ten (40%) had a history of PCP, 13 (53%) had viral enteropathy, and 14 (56%) had growth failure at transplant. Two patients had autoimmune phenomena (1 polyarticular juvenile idiopathic arthritis [JIA]; 1 probable autoimmune hepatitis). Two siblings were affected by macrophage activation syndrome (MAS) prior to transplant (patients 8 and 9). Patient 8 was diagnosed with polyarticular JIA at 14 months of age and treated with etanercept, infliximab, MTX, and local and systemic steroids. The diagnosis of MHC class II deficiency was made following immunological evaluation for prolonged fever at 7 years of age. Three months prior to HCT, she developed MAS with persistent fever, cytopenia, high serum ferritin (highest, 38 348 μg/L), elevated triglycerides, and low fibrinogen. She responded to steroid and CSA. There were no clinical features and no laboratory evidence of MAS reactivation during HCT. Patient 9 was diagnosed at 22 months of age after the diagnosis of MHC class II deficiency was made in patient 8. Patient 9 had chronic diarrhea since birth but did not have history of JIA. Similarly, patient 9 developed MAS while waiting for HCT; she responded well to steroid and CSA. Three patients (patients 15, 19, and 21) had novel homozygous RFXANK mutation c.477C>A (p.Ser159Arg), and patient 7 had novel homozygous CIITA mutation c.3003C>G (p.D1001E).
Transplantation characteristics
Transplantation characteristics are summarized in Tables 2 and 3. The median age at transplant was 21.4 months (range, 0.9 months to 7.8 years) and the median interval between diagnosis and transplant was 9.2 months (range, 0.9 months to 6 years). Six (24%) had MFDs, 12 (48%) had UDs, and 7 (28%) had HIDs. Unmanipulated PBSCs were the stem cell source in 10 (40%) patients, and 7 (28%) received TCRαβ/CD19-depleted parental PBSCs. Only 1 patient received a cord blood transplant. Treosulfan-based RTC was used in 84% of patients (n = 21). Twenty-one patients (84%) received alemtuzumab (n = 14) or ATG (n = 8) as serotherapy.
Engraftment and transplant-related complications
The median times to neutrophil and platelet engraftment were 15 days (range, 8-22) and 16 days (range, 11-42), respectively (Table 3). Thirteen (52%) patients had acute GvHD, of whom 4 (16%) had grade II acute cutaneous GvHD, and none had visceral GvHD. None had grade III-IV acute GvHD or chronic GvHD. All 5 patients who had pretransplant parenteral nutritional–dependent enteropathy were able to establish full enteral nutrition posttransplant. The parenteral nutrition was discontinued at a median of 135 days post-HCT (range, 67-641) in these patients. Sixteen (64%) patients developed new-onset viremia during transplant. Five of 6 deaths were due to transplant-related complications (4 pneumonitis and 1 cerebral hemorrhage). Patient 5 had congenital pelvic ureteric junction obstruction and died of urosepsis after 2 successful MFD transplants and good immune reconstitution.
OS and EFS
The median duration of follow-up of surviving patients was 2.9 years (range, 0.7-7.3). The 3-year OS was 78% (95% CI, 56-91), increasing to 94% (95% CI, 66-99) for children transplanted after 2008 (n = 19) vs 33% (95% CI, 46-68) for the children transplanted between 1995 and 2008 (n = 6; P = .003) (Figure 1A-B). For transplants after 2008, the OS was comparable among MFDs (100%), UDs (100%), and HIDs (83%; 95% CI, 27-97; P = .40) (Figure 1C). Age at transplant (P = .59), stem cell source (P = .50), total nucleated cell dose (P = .59) and CD34 cell dose (P = .59), and stem cell dose were not associated with OS in patients who were transplanted between 2009 and 2018.
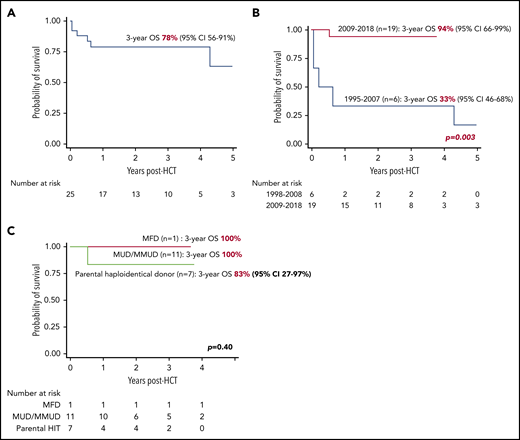
Transplant survival in children with MHC class II deficiency. (A) OS for the entire cohort (n = 25). (B) OS according to years of transplant. (C) OS according to donor type in patients transplanted between 2008 and 2018 (n = 19).
Transplant survival in children with MHC class II deficiency. (A) OS for the entire cohort (n = 25). (B) OS according to years of transplant. (C) OS according to donor type in patients transplanted between 2008 and 2018 (n = 19).
The 3-year EFS for the cohort was 78% (95% CI, 56-91). Of the 3 patients (patients 2, 5, and 22) who had graft failure, 2 died (patients 2 and 22) during second transplant (1 cerebral hemorrhage, 1 pneumonitis). Patient 5 underwent a successful second transplant with good immune reconstitution but died of urosepsis 3.8 years later.
Donor chimerism, immune reconstitution, and long-term disease outcome
Long-term follow-up data, available for 14 transplant survivors, are shown in Table 4. The median age of long-term survivors was 5.3 years (range, 2.2-14), with a median duration of follow-up of 3.5 years (range, 1.1-7.3). The median donor myeloid and lymphocyte chimerism at last follow-up (n = 19) was 100% (range, 0-100) and 100% (range, 64-100), respectively. Immune-reconstitution kinetics within the first 12 months was available for 18 of 19 survivors (1 patient was excluded for on-going active GvHD treatment; Figure 2). Compared with disease controls, patients transplanted for MHC class II deficiency had significantly lower CD4+ T-lymphocyte counts at 5 months (P =.06), 6 months (P = .01), and 12 months (P = .002) posttransplant, as well as at latest follow-up (P = .01) (Figure 3). Although 7 survivors (50%) had low or low-normal CD4+ T lymphocytes at last review, the remainder had CD4+ T-lymphocyte counts in the normal range. There was no significant difference in circulating CD3+ cells (P = .61), CD8+ cells (P = .59), CD19+ cells (P = .90), NK cells (P = .22), or percentage of activated T lymphocytes (HLA-DR) (P = .66) between patients and controls at any time point posttransplant. Donor type had no impact on immune reconstitution kinetics (supplemental Figure 1, available on the Blood Web site). All long-term survivors were well, with no end organ damage (Table 4). Patient 14 had good neurological recovery after enterovirus meningitis with communicating hydrocephalus. All survivors were off Ig replacement and had protective vaccine responses. None had any significant infection or autoimmunity.

Immune reconstitution kinetics posttransplant. Mean CD3+ cells (A), mean CD4+ cells (B), mean CD19 cells (C), mean CD8 cells (D), mean activated T cells (E), and mean NK cells (F) were measured at different time points posttransplant. Disease controls (n = 36) included patients who were transplanted for SCID (n = 10), CD40 ligand deficiency (n = 2), Wiskott-Aldrich syndrome (n = 7), chronic granulomatous disease (n = 13), and other PID (n = 4; 1 NK deficiency, 1 inducible costimulator deficiency, 1 hemophagocytic lymphohistiocytosis, 1 combined immunodeficiency). *P = .06, **P < .05.
Immune reconstitution kinetics posttransplant. Mean CD3+ cells (A), mean CD4+ cells (B), mean CD19 cells (C), mean CD8 cells (D), mean activated T cells (E), and mean NK cells (F) were measured at different time points posttransplant. Disease controls (n = 36) included patients who were transplanted for SCID (n = 10), CD40 ligand deficiency (n = 2), Wiskott-Aldrich syndrome (n = 7), chronic granulomatous disease (n = 13), and other PID (n = 4; 1 NK deficiency, 1 inducible costimulator deficiency, 1 hemophagocytic lymphohistiocytosis, 1 combined immunodeficiency). *P = .06, **P < .05.
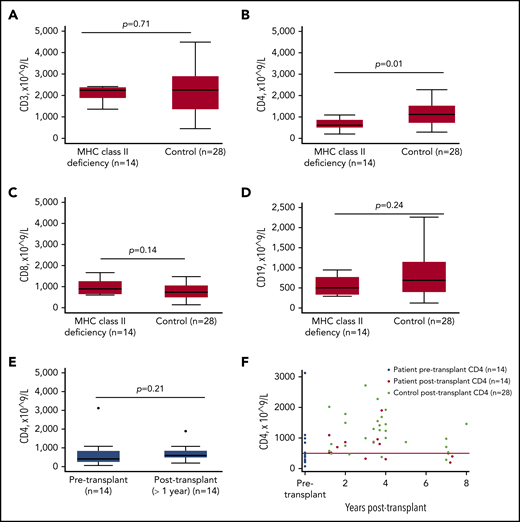
Lymphocyte subsets at latest follow-up (>1 year posttransplant). CD3+ cells (A), CD8+ cells (C), and CD19+ (D) cells were comparable between patients with MHC class II deficiency (n = 14) and controls. (B) CD4+ cells were significantly lower in patients with MHC class II deficiency compared with controls (P = .01). (E) There was no significant difference in CD4+ cells before and after transplant in patients with MHC class II deficiency. (F) Pre- and posttransplant CD4+ cells in individual patients and controls.
Lymphocyte subsets at latest follow-up (>1 year posttransplant). CD3+ cells (A), CD8+ cells (C), and CD19+ (D) cells were comparable between patients with MHC class II deficiency (n = 14) and controls. (B) CD4+ cells were significantly lower in patients with MHC class II deficiency compared with controls (P = .01). (E) There was no significant difference in CD4+ cells before and after transplant in patients with MHC class II deficiency. (F) Pre- and posttransplant CD4+ cells in individual patients and controls.
Discussion
This is one of the largest HCT series for MHC class II deficiency from a single transplant center over the past 2 decades. A number of important observations emerge from this report. Although the majority of patients present with classical clinical and immunological features, some patients can have noninfectious manifestations and an unusual pattern of laboratory results. OS has improved significantly, and no patient had severe acute GvHD or chronic GvHD. Despite numerically decreased CD4+ T-lymphocyte reconstitution, all transplant survivors had a good immunological outcome.
Children with MHC class II deficiency usually present with a clinical phenotype that is very similar to SCID and generally have severe CD4+ T lymphocytopenia, hypogammaglobulinemia, and lack of antigen-specific antibody responses. The CD4+ lymphocytopenia reflects the abnormal CD4+ thymocyte development due to defective MHC class II expression in the thymus. CD8+ T-lymphocyte counts may be normal or raised, leading to an inverted CD4/CD8 ratio. The diagnosis is confirmed by absent or very low HLA-DR expression on lymphocytes, which is the immunological hallmark of the disease. In our cohort, 6 patients (24%) had a normal CD4+ T-lymphocyte count, 4 patients (16%) had a high CD8+ T-lymphocyte count, and 8 patients (32%) had a normal CD4/CD8 ratio. Autoimmunity was the first presentation in patient 8, who was treated with multiple biologic agents for refractory arthritis before the diagnosis of MHC class II deficiency was made. She and her younger sibling (patient 9), both of whom had homozygous CIITA mutation, developed macrophage activation syndrome prior to transplant, which has not been reported previously. Although disseminated BCGosis (disseminated infection) is not expected in these patients because of the presence of residual immunity in the form of CD8+ T lymphocytes and NK cells, 1 patient in our cohort had disseminated BCGosis prior to transplant.
We have shown that the 3-year OS has improved to 94% for children with MHC class II deficiency transplanted since 2008, which is comparable to HCT for children with severe combined immunodeficiency.3,6-10 Small et al reported transplant survival of 69% in 16 children with MHC class II deficiency from 5 transplant centers in Europe and the United States.11 Al-Mousa et al12 reported a similar transplant survival of 66% in a cohort of 30 patients from a single transplant center in Saudi Arabia. Many factors have contributed to this improvement in Great North Children's Hospital, including a detailed graft selection hierarchy, superior HLA-matching technology, improved methods for graft manipulation, improved supportive care, vigilant infection surveillance and preemptive treatment, more effective antimicrobial therapy, and a multidisciplinary team approach prior to and after HCT. In our center, we have used RTC with fludarabine, treosulfan, and alemtuzumab for family and UD transplants since 2009. Our haploidentical transplant strategy in this cohort includes conditioning with fludarabine, treosulfan, thiotepa, ATG (Grafalon), and rituximab and graft manipulation by TCRαβ/CD19 depletion of PBSCs. The OS following haploidentical transplant in our cohort was comparable with family and UD transplants, with a single fatality. Despite being diagnosed at 6 months of age, the deceased patient was referred for transplant at 6 years, by which time he had multiple viral infections, gut failure, and chronic lung disease. The use of alemtuzumab and graft engineering has reduced the incidence of acute GvHD in our cohort compared with the high reported incidence ranging from 47% to 73%.9,12,13 A high incidence of acute GvHD in previous studies may be due to inadequate in vivo T-lymphocyte depletion and chronic viral infection. Despite the fact that pre-HCT enteropathy is common in our study cohort, no patient had gut GvHD.
The long-term disease outcome in our cohort is excellent, with good immunological outcomes, no infections, and recovery of previous organ damage. In contrast to Klein et al’s observation of persistent CD4+ lymphopenia in all 8 transplant survivors in their series of 19 transplants, 50% (n = 7) of the survivors have normal CD4+ lymphocytes posttransplant.7 Godthelp et al described an impaired immune repertoire in 2 patients with partial engraftment post-HCT.14 The persistent CD4+ T lymphopenia in some patients might be explained by impaired thymocyte maturation caused by defective MHC class II expression on thymic epithelia. Our cohort with the largest series of transplant survivors showed that, despite low CD4+ T-lymphocyte numbers, transplanted patients exhibit normalization of antigen-specific T-lymphocyte stimulation and antibody production in response to immunization antigens. However, the long-term impact of decreased CD4+ T-lymphocyte reconstitution is unclear, particularly the risks of autoimmunity and premature immunosenescence.
In conclusion, HCT is a safe curative therapy for children with MHC class II deficiency. Unrelated and parental HIDs are associated with excellent survival at Great North Children's Hospital, comparable to MFDs. Because young age at HCT is associated with a favorable outcome in children with MHC class II deficiency, HCT should be performed as early as possible after the diagnosis, before the onset of disease-related organ damage.7 Family screening plays an important role in advancing the transplant care for children with MHC class II deficiency. There is a need for a multicenter study using registry data to delineate the predictors of transplant survival and long-term disease outcomes in children with MHC class II deficiency. Advances in graft manipulation, as well as additional cellular therapy and patient-tailored conditioning regimens, may enable precise personalized transplant care and further improve transplant survival while reducing late effects, such as infertility. In addition, it is conceivable that, in the future, gene therapy may offer an alternative option for children with MHC class II deficiency.
For original data, please contact Su Han Lam (s.lum@nhs.net).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all patients and their families, as well as the treating multidisciplinary team.
Authorship
Contribution: S.H.L. collected data, performed the statistical analysis, interpreted data, and prepared the manuscript; M.S. and A.R.G. conceptualized the research, performed statistical analysis, interpreted data, and wrote and critically reviewed the manuscript; C.A., P.M., B.M., H.W., B.A.-S., D.A.-Z., H.A.-M., W.A.-H., H.A.A., B.S., S. Habibollah, R.M., and L.G. collected the data and reviewed the manuscript; A.F., E.W., K.E., and M.E. reviewed the manuscript; and T.F., S.O., Z.N., S. Hambleton, A.C., and M.A. critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Su Han Lum, Clinical Resource Building, Floor 4, Block 2, Great North Children’s Hospital, Queen Victoria Rd, Newcastle upon Tyne NE1 4LP, United Kingdom; e-mail: s.lum@nhs.net.


