

Key Points
H499C and H499Y mutations enhance activation of TpoR by novel L498W and canonical S505N mutations.
Activation of TpoR by eltrombopag and the L498W, S505N, and W515K mutants depends on W491, which might be accessible on the cell surface.

Abstract
Mutations in the MPL gene encoding the human thrombopoietin receptor (TpoR) drive sporadic and familial essential thrombocythemias (ETs). We identified 2 ET patients harboring double mutations in cis in MPL, namely, L498W-H499C and H499Y-S505N. Using biochemical and signaling assays along with partial saturation mutagenesis, we showed that L498W is an activating mutation potentiated by H499C and that H499C and H499Y enhance the activity of the canonical S505N mutation. L498W and H499C can activate a truncated TpoR mutant, which lacks the extracellular domain, indicating these mutations act on the transmembrane (TM) cytosolic domain. Using a protein complementation assay, we showed that L498W and H499C strongly drive dimerization of TpoR. Activation by tryptophan substitution is exquisitely specific for position 498. Using structure-guided mutagenesis, we identified upstream amino acid W491 as a key residue required for activation by L498W or canonical activating mutations such as S505N and W515K, as well as by eltrombopag. Structural data point to a common dimerization and activation path for TpoR via its TM domain that is shared between the small-molecule agonist eltrombopag and canonical and novel activating TpoR mutations that all depend on W491, a potentially accessible extracellular residue that could become a target for therapeutic intervention.
Introduction
Patients with the polycythemia vera, essential thrombocythemia (ET), or primary myelofibrosis (PMF) type of myeloproliferative neoplasm harbor driver mutations in JAK2, CALR, or MPL.1 Mutations in the human thrombopoietin receptor (TpoR) are restricted to ET and PMF, largely W515K/L/A/R substitutions in the amphipathic juxtamembrane (JM) domain2,3 and more rarely S505N in the transmembrane (TM) domain.4,5 These mutations lead to ligand-independent receptor dimerization and resultant activation of signaling via JAK2.6-8 Although 20% of ET and 10% to 15% of PMF patients harbor none of the 3 driver mutations, some of these triple-negative (TN) patients may harbor noncanonical MPL mutations.6,9-11
We analyzed 2 patients with ET negative for JAK2, CALR, and MPL W515 mutations. We found that each patient harbored double cis mutations in MPL, namely, L498W-H499C and H499Y-S505N. These mutations have never been described in such patients, and both combinations were active. Using structure-guided mutagenesis, dimerization, biochemical, and signaling assays, we defined a broad mechanism of activation shared by these new mutants, as well as by the small-molecule agonist eltrombopag and the canonical S505N and W515 mutants. This mechanism depends on a potentially accessible region upstream of the JM-TM region.
Study design
Hemagglutinin-tagged human TpoR wild-type (WT) and mutant complementary DNAs, as well as a TM-intracellular (TM-IC) TpoR constructs lacking the extracellular domain,12 were cloned into the pMX-IRES-GFP bicistronic vector. Transcriptional basal and ligand-dependent activation of STAT5 downstream of TpoR was tested in γ2A and HEK-293T cells in a dual-luciferase reporter assay (DualGlo; Promega, Madison, WI).12 Cytokine dependency of proliferation in stably transduced Ba/F3 cells was determined as previously described.12 The levels of TpoR total protein expression were determined by western blotting using antihemagglutinin antibodies (Roche Applied Science) or anti-TpoR antibodies (MilliporeSigma, Burlington, MA). Cell-surface localization of TpoR was evaluated by flow cytometry using anti-TpoR(CD110) phycoerythrin antibodies (Clone REA250; Miltenyi Biotec, Bergisch Gladbach, Germany). TpoR dimerization was assessed in HEK-293T cells using the NanoBiT complementation assay.13,14
MPL sequencing in the 2 study patients was performed with a diagnostic goal, with patient approval according to local French and European guidelines.
Results and discussion
The TpoR L498W-H499C variant was identified by targeted sequencing in a patient with TN ET. Whole-exome sequencing of genomic bone marrow DNA did not identify any other potential driver when assessing a 171-gene myeloid pathology panel. Targeted next-generation sequencing to assess variant allele frequency in the patient’s peripheral whole mononucleated cells (18%) and granulocytes (43%) pointed toward an acquired mutation (supplemental Table 1). Both single mutations led to ligand-independent STAT5 transcriptional activity, although L498W had a stronger effect, and the combination was even stronger (Figure 1A). When stably transduced and at equivalent levels in the interleukin-3–dependent Ba/F3 cell line, autonomous growth was detected in L498W, which was enhanced in L498W-H499C (Figure 1B). Cell-surface localization of L498W was similar to that of the WT receptor (Figure 1C).
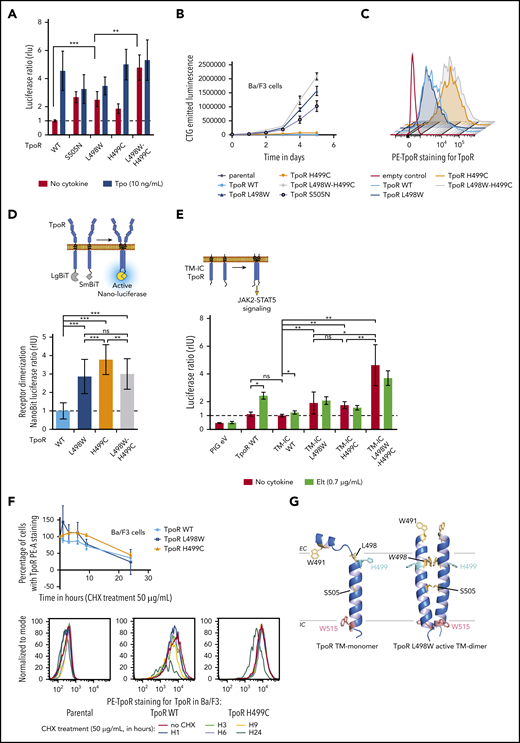
Activation of TpoR signaling by L498W-H499C mutations. (A) Effects of H499C and L498W mutations on TpoR signaling. HEK-293T cells were transiently transfected with the indicated hemagglutinin (HA)-TpoR variants, along with complementary DNAs (cDNAs) coding for JAK2, STAT5, and SpiLuc Firefly luciferase reporter reflecting STAT5 transcriptional activity and normalized with a control reporter (pRL-TK) containing Renilla luciferase. Shown are averages of 3 independent experiments, each performed in triplicate + standard deviation (SD; error bars). (B) Induction of autonomous proliferation of Ba/F3 cells by L498W and L498W-H499C mutants of TpoR. Ba/F3 cells stably expressing the TpoR mutants of interest were grown in the absence of cytokine. Cell proliferation was assessed by CellTiter-Glo (CTG) luminescent cell viability assay. (C) H499C increased cell-surface localization of TpoR. HEK-293T cells were transiently transfected with the indicated HA-TpoR variants. TpoR cell-surface localization among fluorescein isothiocyanate (FITC)–positive cells was assessed by flow cytometry using anti-TpoR antibody. Shown is 1 representative of 2 experiments. (D) TpoR dimerization in the absence of cytokine. Dimerization of TpoR was determined in HEK-293T using the NanoBiT luciferase protein fragment complementation assay using Nano-luciferase fragments fused in frame with the cytosolic domain of TpoR. (E) The extracellular domain of TpoR was not needed for constitutive activation by the L498W-H499C mutant. HEK-293T cells were transiently transfected with the indicated HA-TM-intracellular (TM-IC) TpoR variants, along with cDNAs coding for JAK2, STAT5, and SpiLuc Firefly luciferase reporter reflecting STAT5 transcriptional activity and normalized with a control reporter (pRL-TK) containing Renilla luciferase. Shown are averages of 4 independent experiments, each performed in triplicate + SD (error bars). (F) TpoR H499C continued to be overexpressed after cycloheximide (CHX) treatment. Ba/F3 cells stably expressing the TpoR mutants of interest were grown in presence of interleukin-3 and treated with CHX (50 μg/mL). TpoR cell-surface localization among FITC+ cells was assessed by flow cytometry using anti-TpoR antibody. Upper panel is an average of 3 independent experiments + SD (error bars). Lower panel is 1 representative of 3 experiments. (G) Model of the monomeric WT and dimeric L498W TM domains (TMDs) of the TpoR. On the left, the monomeric TMD has a break in the helical secondary structure in the region N-terminal to H499.12 On the left, in the L498W-active TMD dimer, the helix is induced in the region N-terminal to H499. *P < .05, **P < .01, ***P < .001. EC, extracellular; Elt, eltrombopag; eV, empty vector; LgBit, Nano-luciferase large-bit subunit; ns, nonsignificant (nonparametric multiple-comparison Steel test with control); PE, phycoerythrin; PIG, pMX-IRES-GFP vector; SmBit, Nano-luciferase small-bit subunit.
Activation of TpoR signaling by L498W-H499C mutations. (A) Effects of H499C and L498W mutations on TpoR signaling. HEK-293T cells were transiently transfected with the indicated hemagglutinin (HA)-TpoR variants, along with complementary DNAs (cDNAs) coding for JAK2, STAT5, and SpiLuc Firefly luciferase reporter reflecting STAT5 transcriptional activity and normalized with a control reporter (pRL-TK) containing Renilla luciferase. Shown are averages of 3 independent experiments, each performed in triplicate + standard deviation (SD; error bars). (B) Induction of autonomous proliferation of Ba/F3 cells by L498W and L498W-H499C mutants of TpoR. Ba/F3 cells stably expressing the TpoR mutants of interest were grown in the absence of cytokine. Cell proliferation was assessed by CellTiter-Glo (CTG) luminescent cell viability assay. (C) H499C increased cell-surface localization of TpoR. HEK-293T cells were transiently transfected with the indicated HA-TpoR variants. TpoR cell-surface localization among fluorescein isothiocyanate (FITC)–positive cells was assessed by flow cytometry using anti-TpoR antibody. Shown is 1 representative of 2 experiments. (D) TpoR dimerization in the absence of cytokine. Dimerization of TpoR was determined in HEK-293T using the NanoBiT luciferase protein fragment complementation assay using Nano-luciferase fragments fused in frame with the cytosolic domain of TpoR. (E) The extracellular domain of TpoR was not needed for constitutive activation by the L498W-H499C mutant. HEK-293T cells were transiently transfected with the indicated HA-TM-intracellular (TM-IC) TpoR variants, along with cDNAs coding for JAK2, STAT5, and SpiLuc Firefly luciferase reporter reflecting STAT5 transcriptional activity and normalized with a control reporter (pRL-TK) containing Renilla luciferase. Shown are averages of 4 independent experiments, each performed in triplicate + SD (error bars). (F) TpoR H499C continued to be overexpressed after cycloheximide (CHX) treatment. Ba/F3 cells stably expressing the TpoR mutants of interest were grown in presence of interleukin-3 and treated with CHX (50 μg/mL). TpoR cell-surface localization among FITC+ cells was assessed by flow cytometry using anti-TpoR antibody. Upper panel is an average of 3 independent experiments + SD (error bars). Lower panel is 1 representative of 3 experiments. (G) Model of the monomeric WT and dimeric L498W TM domains (TMDs) of the TpoR. On the left, the monomeric TMD has a break in the helical secondary structure in the region N-terminal to H499.12 On the left, in the L498W-active TMD dimer, the helix is induced in the region N-terminal to H499. *P < .05, **P < .01, ***P < .001. EC, extracellular; Elt, eltrombopag; eV, empty vector; LgBit, Nano-luciferase large-bit subunit; ns, nonsignificant (nonparametric multiple-comparison Steel test with control); PE, phycoerythrin; PIG, pMX-IRES-GFP vector; SmBit, Nano-luciferase small-bit subunit.
We and others had previously shown that the human TpoR is mainly a cell-surface monomer that dimerizes, in the presence of either Tpo or the small-molecule agonist eltrombopag12,15 or when mutated (S505N, W515K), via a structural change in the region around H499. We hypothesized that L498W and H499C would induce persistent dimerization of TpoR. As detected by protein complementation using the LargeBit and SmallBit fragments of Nano-luciferase,13,14 each mutation induced strong dimerization of the TpoR cytosolic domains (Figure 1D; supplemental Figure 1B).
Although TpoR H499C did not support proliferation in Ba/F3 cells, its dimerization was comparable to or higher than that of L498W (Figure 1D). Cell-surface localization and stability of H499C were increased in comparison with WT or L498W (Figure 1C,F; supplemental Figure 1E-F). Total expression of the mutants was comparable across cell lines (supplemental Figure 2A). These data suggest that H499C, although it increases cell-surface TpoR localization, dimerizes TpoR in a conformation not optimal for signaling. Indeed, different dimeric orientations are compatible with different degrees of signaling.16,17
To assess whether dimerization and activation involve only the TM intracellular domains or require the extracellular region as well, we truncated TpoR upstream of position 489 and introduced the mutations into this receptor construct.13 Both the single and double mutants exhibited significantly more signaling than the WT truncated receptor (Figure 1E; supplemental Figure 2C). Thus, the mutations act on the TM cytosolic domains. In contrast, treatment with eltrombopag activated the TpoR WT but not the mutants, in agreement with a requirement for extraamino acid residues upstream of H499.15
We next asked whether other residues could activate at position 498 and whether Trp could activate at other positions between residues 494 and 504. We found that activation was unique to Trp at 498 (supplemental Figures 1C-D and 2B). On the basis of previous structural data showing that dimerization induces a helical conformation around H499,7,12 L498W is predicted to stabilize a TM dimer with W498 facing partially toward the dimer interface, as does S505N (Figure 1G; supplemental Figure 1A).
In a second ET patient, we identified the double mutation H499Y-S505N (in cis) by targeted sequencing (supplemental Table 1). Although the H499Y mutant was found to be active alone, the double mutant exhibited significantly enhanced activation of STAT5 over S505N (Figure 2A). Both mutants were localized on the cell surface and in the cells at similar or higher levels than the WT receptor (Figure 2B; supplemental Figure 2D). Examining whether residues other than H499C/H499Y could activate at position 499, we found that it was not the case for Leu, Phe, Trp, or Asn (Figure 2C; supplemental Figure 2E), a finding that is corroborated by a recent study.18 The next question was whether these H499 mutations also enhanced signaling by other TpoR mutants such as L498W or W515K. H499C and H499Y increased activity of both S505N and L498W, but not W515K (Figure 2D). The enhancement was specific for the H499 mutations, because L498W failed to enhance S505N or W515K signaling (Figure 2D). That H499 mutations did not alter W515K activity might reflect different active dimeric interfaces adopted by S505N/L498W vs W515K.

H499 and W491 play opposite roles in regulating activation of TpoR by several activating mutations. (A) Effect of TpoR H499Y mutation on constitutive activity of the S505N mutant. (B) H499Y and H499Y-S505N increase TpoR cell-surface localization. (C) Effects of aromatic and aliphatic residues at H499 on TpoR activity. (D) Effects of H499Y/H499C on the constitutive activities of the indicated TpoR mutants (upper) and on their cell-surface localization (lower). (E) Model of the dimeric active S505N and inactive W491A TM domains of the TpoR. On the left, in the active S505N mutant, the N505 residues face one another. On the right, in the inactive W491A mutant, the H499 residues face one another and would therefore be inaccessible to eltrombopag (Elt). (F) Effects of W491A/W491K mutations on the indicated TpoR mutants and on stimulations by Tpo or Elt. In panels A, C, D, and F, HEK-293T cells were transiently transfected with the indicated hemagglutinin (HA)-TpoR variants, along with complementary DNAs coding for JAK2, STAT5, and SpiLuc Firefly luciferase reporter reflecting STAT5 transcriptional activity and normalized with a control reporter (pRL-TK) containing Renilla luciferase. Shown are averages of 3 to 4 independent experiments, each performed in triplicate + standard deviation (error bars). In panels B and D, HEK-293T cells were transiently transfected with the indicated HA-TpoR variants. TpoR cell-surface localization among fluorescein isothiocyanate–positive cells was assessed by flow cytometry using anti-TpoR antibody. Shown is 1 representative of 2 experiments. **P < .01, ***P < .001. EC, extracellular; eV, empty vector; ns, nonsignificant (nonparametric multiple-comparison Steel test with control); PE, phycoerythrin; PIG, pMX-IRES-GFP vector.
H499 and W491 play opposite roles in regulating activation of TpoR by several activating mutations. (A) Effect of TpoR H499Y mutation on constitutive activity of the S505N mutant. (B) H499Y and H499Y-S505N increase TpoR cell-surface localization. (C) Effects of aromatic and aliphatic residues at H499 on TpoR activity. (D) Effects of H499Y/H499C on the constitutive activities of the indicated TpoR mutants (upper) and on their cell-surface localization (lower). (E) Model of the dimeric active S505N and inactive W491A TM domains of the TpoR. On the left, in the active S505N mutant, the N505 residues face one another. On the right, in the inactive W491A mutant, the H499 residues face one another and would therefore be inaccessible to eltrombopag (Elt). (F) Effects of W491A/W491K mutations on the indicated TpoR mutants and on stimulations by Tpo or Elt. In panels A, C, D, and F, HEK-293T cells were transiently transfected with the indicated hemagglutinin (HA)-TpoR variants, along with complementary DNAs coding for JAK2, STAT5, and SpiLuc Firefly luciferase reporter reflecting STAT5 transcriptional activity and normalized with a control reporter (pRL-TK) containing Renilla luciferase. Shown are averages of 3 to 4 independent experiments, each performed in triplicate + standard deviation (error bars). In panels B and D, HEK-293T cells were transiently transfected with the indicated HA-TpoR variants. TpoR cell-surface localization among fluorescein isothiocyanate–positive cells was assessed by flow cytometry using anti-TpoR antibody. Shown is 1 representative of 2 experiments. **P < .01, ***P < .001. EC, extracellular; eV, empty vector; ns, nonsignificant (nonparametric multiple-comparison Steel test with control); PE, phycoerythrin; PIG, pMX-IRES-GFP vector.
In the active S505N dimer, Asn was in the dimer interface of the coiled coil formed by the 2 TM domains, whereas H499 was oriented away from the interface12 (supplemental Figure 1A lower). We previously found that substitution of H499 with Leu (the WT residue in murine TpoR) in the TM-intracellular construct of TpoR induced dimerization, but in an inactive orientation.12 The unique behavior of H499Y and H499C suggests that these substitutions destabilize the inactive orientation of the WT receptor (as in H499L) and allow the TM helices to form more stabilizing interhelical contacts in the active dimer.
On the basis of structural and mutational data,7,12 we modeled the active TpoR dimeric state using an α-helical structure in the region of H499. Another Trp residue (W491) exists upstream on the same helical face as W498 and S505 (both predicted to occupy an “a” position in a left-handed coiled coil motif; Figures 1G and 2E; supplemental Figure 1A), 25 residues from W515 of the RWQFP insert. The W515 position also belongs to the same face, but in a “d” position (supplemental Figure 1A).7,19 We hypothesized that, when rotated inward, the W491 residue might stabilize the active state promoted by other mutations (L498W, S505N, and W515K). Given the size of a Trp in the interface, it could push apart helices, which is necessary to allow crossing at S505, leading to activation.12 Indeed, replacing W491 with A or K prevented TpoR activation by L498W, S505N, and W515K (Figure 2F), although neither cell-surface nor global expression was altered (Figure 2F; supplemental Figure 2F). Thus, W491 is crucial for the changes in tilt and orientation required for activation.12
We also tested whether W491 is required, like H499, for activation by eltrombopag. W491 mutations impaired eltrombopag-mediated activation (Figure 2F) but did not block Tpo-induced activation. We and others showed by polarized infrared spectroscopy and nuclear magnetic resonance studies that eltrombopag induces helix formation around H499 and TpoR dimerization.12,15 We propose now that eltrombopag activates TpoR by breaking TpoR-membrane interactions via binding to both W491 and H499 and shares the path of activation with the canonical mutants.
Furthermore, our model in Figure 2E predicts that rotating S505 and W515 inward and H499 outward will result in activation stabilized by W491. W491 might potentially be accessible on the extracellular JM domain to become a target for small molecules or antibody-mediated inhibition of TpoR. Other mutants such as those described here are indeed likely to be identified in TN ET patients, and they are likely to depend on W491. Interestingly, a recent study using deep mutagenesis and sequencing reported that mutations at positions 490 and 492 also lessened the activity of the S505N mutant.18 This entire region might thus be important in the activation of TpoR.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Lidvine Genet and Céline Mouton for expert technical support.
G.L. was supported by a fellowship awarded from the Foundation “Les Avions de Sébastien.” L.N.V. has received support from the de Duve Institute Maurange Funds postdoctoral fellowship and the MOVE-IN Louvain fellowship. S.N.C. is Honorary Research Director at Fonds de la Recherche Scientifique (FRS-FNRS) Belgium and has received funds from Ludwig Institute for Cancer Research, Fondation contre le Cancer, Salus Sanguinis, and Action de Recherche Concertée Projects 16/21-073 and WelBio F 44/8/5-MCF/UIG-10955.
Authorship
Contribution: G.L., J.-P.D., and I.C. planned the study strategy, performed experiments, interpreted data, and wrote the paper; E.L. and L.N.V. planned specific experiments and interpreted data; S.C., B.P., B.C., and J.-M.Z. provided data on patients; S.O.S. provided structural models and protein structure predictions; and S.N.C. planned research, interpreted data, and wrote the paper.
Conflict-of-interest disclosure: S.N.C. is cofounder of MyeloPro Research and Diagnostics GmbH, Vienna, Austria. The remaining authors declare no competing financial interests.
Correspondence: Stefan N. Constantinescu, Ludwig Institute for Cancer Research and de Duve Institute, Université Catholique de Louvain, Avenue Hippocrate 75, 1200 Woluwe-Saint-Lambert, Brussels, Belgium; e-mail: stefan.constantinescu@bru.licr.org.
