Key Points
Type 1 IFN signaling is critical for the development of DIC in endotoxemia and bacterial sepsis.
Type 1 IFN signaling mediates the release of HMGB1, which promotes DIC by inducing phosphatidylserine exposure.

Abstract
Bacterial infection not only stimulates innate immune responses but also activates coagulation cascades. Overactivation of the coagulation system in bacterial sepsis leads to disseminated intravascular coagulation (DIC), a life-threatening condition. However, the mechanisms by which bacterial infection activates the coagulation cascade are not fully understood. Here we show that type 1 interferons (IFNs), a widely expressed family of cytokines that orchestrate innate antiviral and antibacterial immunity, mediate bacterial infection–induced DIC by amplifying the release of high-mobility group box 1 (HMGB1) into the bloodstream. Inhibition of the expression of type 1 IFNs and disruption of their receptor IFN-α/βR or downstream effector (eg, HMGB1) uniformly decreased gram-negative bacteria-induced DIC. Mechanistically, extracellular HMGB1 markedly increased the procoagulant activity of tissue factor by promoting the externalization of phosphatidylserine to the outer cell surface, where phosphatidylserine assembles a complex of cofactor-proteases of the coagulation cascades. These findings not only provide novel insights into the link between innate immune responses and coagulation, but they also open a new avenue for developing novel therapeutic strategies to prevent DIC in sepsis.
Introduction
Sepsis, an infection-induced critical illness characterized by multiple organ dysfunctions, is a leading cause of hospital mortality often accompanied by coagulopathy.1,2 The most severe form of coagulopathy is known as disseminated intravascular coagulation (DIC), which is manifested by systemic activation of the coagulation cascades, deposition of fibrin, and aggregation of platelets in the microvasculature throughout the body.2 This pathogenic process compromises the blood supply to various organs, thereby promoting multiple organ dysfunctions.2,3 DIC could also lead to the excessive consumption of coagulation factors and platelets, resulting in pronounced bleeding from various sites. The occurrence of DIC significantly contributes to mortality in patients with sepsis.3,4 Although treatment with anticoagulant therapy after the onset of sepsis did not improve the overall outcome in several clinical trials,5-7 pretreatment with heparin, a well-known anticoagulant, significantly ameliorated organ injury and septic lethality in preclinical settings.8
Bacterial endotoxin (lipopolysaccharide [LPS]), the major cell wall component of gram-negative bacteria, could stimulate monocytes or vascular endothelial cells to express tissue factor (TF), a transmembrane glycoprotein that activates the extrinsic coagulation cascade.9-11 Mice with reduced or depleted TF expression in myeloid cells have markedly reduced activation of coagulation in endotoxemia.12 Similarly, pharmacologic suppression of TF activity with neutralizing antibodies could also prevent LPS-induced DIC.12,13 The biological activities of TF are regulated posttranscriptionally by multiple mechanisms, including the availability of the cell surface procoagulant phosphatidylserines (PS) and thiol-disulfide exchange.14,15 Under physiological conditions, most of the PS locate in the inner layer of the cytoplasmic membrane,14 but their externalization markedly increases the procoagulant activity of TF.10,11,14-16 As the common cause of DIC,2 gram-negative sepsis induces robust PS externalization in peripheral leukocytes.16 Pharmacologic blockade of cell surface PS or its protein disulfide isomerase–mediated thiol-disulfide exchange significantly attenuates gram-negative sepsis-induced DIC. Recent advances show that protein disulfide isomerase–mediated thiol-disulfide exchange is also critical for TF activation.15,17 Although it is known that bacterial infection and LPS induce the expression of TF,11 the underlying mechanisms by which gram-negative bacteria or LPS promote TF activation through PS externalization or thiol-disulfide exchange remain largely unknown.
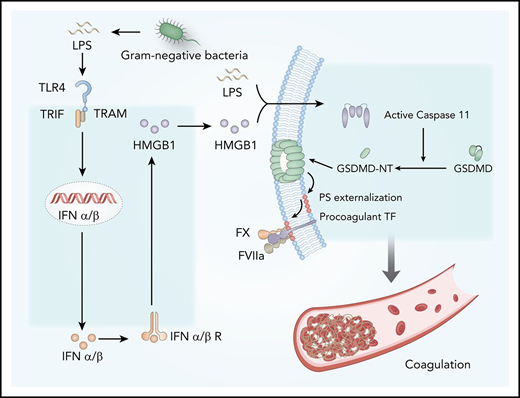
Type 1 interferons (IFNs) are widely expressed cytokines that are readily induced in response to a variety of viral and bacterial infections and shape the antimicrobial innate immune responses.18 Type 1 IFNs, including IFN-ɑ1-4 and IFN-β, signal through their cell surface receptor IFN-α/βR to activate the JAK-STAT pathway, resulting in STAT1-STAT2 dimer formation and nuclear translocation.18,19 This action leads to the upregulated expression of IFN-stimulated genes, which work in concert to confer resistance to viral infection. In addition to viral nucleic acids, gram-negative bacteria or LPS could also stimulate the expression of type 1 IFNs through Toll-like receptor 4 (TLR4) and TIR-domain–containing adapter-inducing interferon-β (TRIF), an adaptor molecule downstream of TLR4.20 Although it is well known that type 1 IFNs are the first line of defense against viral infection,18,19 the roles of type 1 IFNs in gram-negative bacterial infection–induced DIC remain largely unknown. Here we show that type 1 IFN signaling mediates gram-negative bacteria or LPS-induced coagulation by amplifying the release of effectors such as the high-mobility group box 1 (HMGB1) into the extracellular space. Blocking this pathway by genetic or pharmacologic approaches prevents the development of DIC in gram-negative sepsis or endotoxemia. Mechanistically, extracellular HMGB1 markedly increases the procoagulant activity of TF, at least in part, by promoting the exposure of PS on the cell surface, which is important for the assembly of cofactor-protease complexes of the coagulation cascades.11,14
Methods
Mice
The C57BL/6J (wild-type [WT])/Alb-cre+ /Lyz2-cre+/IFN-α/βR1 knockout (KO)/TRIF KO/myeloid differentiation primary response 88 (MyD88) KO mice were purchased from The Jackson Laboratory. Caspase-11 KO, Hmgb1fl/fl, and Pf4-Cre+ mice were provided by T.R.B. and generated as previously described.21,22 Gasdermin D (GSDMD) KO and Vav-cre+ mice were gifts from Professor Jiahuai Han and generated as described elsewhere.23,24 To conditionally knock out HMGB1 in myeloid cells, hepatocytes, hematopoietic progenitor cells, and platelets, Hmgb1fl/fl mice were crossed with Lyz2-cre+, Alb-cre+, Vav-cre+, or Pf4-Cre+ mice, respectively, to generate Hmgb1fl/fl Lyz2-cre+, Hmgb1fl/fl Alb-cre+, Hmgb1fl/fl Vav-cre+, and Hmgb1fl/fl Pf4-cre+ mice. Hepatitis C virus (HCV) and low TF mice25 were donated by Professor Nigel Mackman. More detailed information about the mice is provided in the supplemental Methods (available on the Blood Web site). All experimental animal procedures were approved by the Institutional Animal Care and Use Committees of Central South University.
Antibodies and reagents
Endotoxemia and sepsis model
To induce a LPS-sensitive model,28,29 25 to 30 g mice were primed with 0.4 mg/kg of LPS (intraperitoneal injection) for 7 hours followed by 10 mg/kg of LPS for 8 hours. Macrophages were depleted before LPS injection as previously described.30,31 In the intravital microscopy experiments, mice were injected with 4 mg/kg of LPS for 6 hours to ensure that most blood vessels were not completely occluded and could be better observed, as reported elsewhere.32 Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) as previously described.33 Mice were euthanized at 16 hours after CLP for subsequent experiments. Detailed information, including enzyme-linked immunosorbent assay and histology, is provided in the supplemental Methods.
Intravital microscopy and image analysis
The mouse liver and lung for intravital microscopy were prepared as previously described.34,35 The thrombin substrate (2 μL/mouse), AF647-conjugated anti-mouse fibrin antibody (4 μL/mouse), AF647/anti-CD49b antibody (2 μg/mouse), and AF647-conjugated anti-mouse albumin antibody (0.05 μg/mouse) were injected. Fluorescent images of livers and lungs were photographed by using spinning disk confocal intravital microscopy (SD-IVM) and the Olympus Multiphoton Laser Scanning Confocal Intravital Microscopy, respectively. Images were analyzed as previously described.36 Specific details are provided in the supplemental Methods.
FeCl3-induced thrombosis model
FeCl3-induced mesenteric artery thrombosis was conducted as previously reported.37 The images were captured by using a Nikon fluorescence microscope. Specific details are provided in the supplemental Methods.
Western blot and quantitative reverse transcription polymerase chain reaction
The membranes were incubated with anti-mouse TF antibody (1:1000) and β-actin (1:5000) at 4°C overnight. Quantitative reverse transcription polymerase chain reaction was performed in a LightCycler 480 (Roche) system.
Macrophage cultures and stimulation
Mouse peritoneal macrophages were collected as previously described38 (specific details are described in the supplemental Methods) and plated in 12- or 96-well plates or 6-well slides overnight. Cells were then stimulated with ultrapure LPS (1 μg/mL) or recombinant (r)HMGB1 (400 ng/mL) or ultrapure LPS plus rHMGB1 (premixed for 20 minutes at room temperature) in the presence or absence of milk fat globule epidermal growth factor 8 (MFG-E8) (0.01-0.05 μg/mL) or crude LPS (1 μg/mL) for different hours.
TF activity assay
Macrophages of HCV mice were plated in 96-well plates, incubated with the aforementioned stimuli, and then washed 3 times with phosphate-buffered saline. A 70 μL assay mix containing 10 μL of factor VII and 10 μL of factor X was added into the live cell wells and incubated at 37°C for 30 minutes. Finally, 20 μL of factor Xa substrate was added. The absorbance was recorded immediately at 405 nm every 5 minutes for 25 minutes, and cell surface TF activity was calculated according to the standard curve.
Detection of PS exposure
PS exposure was detected by using immunofluorescence and flow cytometry. Details are described in the supplemental Methods.
Statistical analysis
Data are expressed as the mean ± standard error of the mean. Statistical analyses were performed by using GraphPad Prism 7.0 software. A 2-tailed Student t test was used to compare the differences between 2 groups. When >2 groups were compared, 1-way analyses of variance or 2-way analyses of variance with Bonferroni’s post hoc test were used. The log-rank test was used to analyze survival data. A value of P < .05 was considered statistically significant.
Results
Type 1 IFN signaling mediates the activation of coagulation cascades in endotoxemia
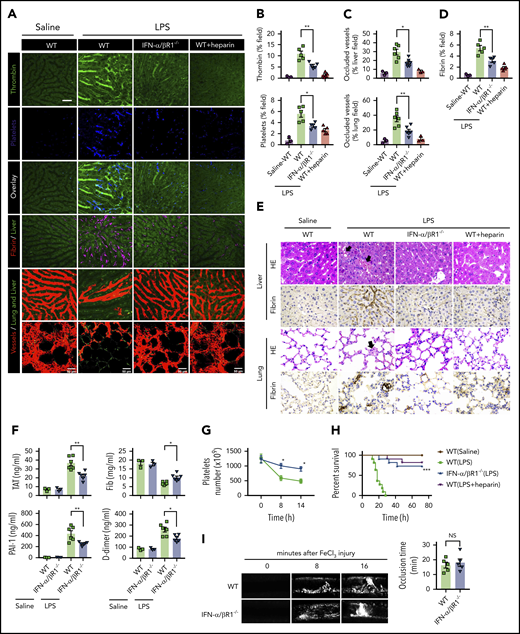
To determine the role of type 1 IFN signaling in the activation of coagulation cascades in endotoxemia, we introduced the internally quenched 5-FAM/QXL-520 FRET substrate of thrombin into the circulation of WT mice or IFN-α/βR1–deficient mice. When cleaved by endogenous active thrombin, green fluorescence signal is produced within the vasculature and can be detected by using intravital microscopy during endotoxemia. Notably, genetic deletion of IFN-α/βR1 almost completely blocked the generation of intravascular thrombin throughout the liver microvasculature (Figure 1A-B). We also observed that the aggregation of platelets, fibrin deposition, and occlusion of the microcirculation were reduced in IFN-α/βR1–deficient mice or heparin pretreated mice compared with that of their WT controls (Figure 1A-D). Accordingly, fibrin deposition in the liver and lung during endotoxemia were also revealed by immunohistochemical staining (Figure 1E). Excessive activation of the coagulation cascades results in an increased consumption of fibrinogen and elevated production of D-dimer from the plasmin-mediated fibrin degradation.4 Markers of DIC include thrombin–antithrombin (TAT) complexes formed during activation of coagulation, plasminogen activator inhibitor type 1 (PAI-1), an inhibitor of fibrinolysis, and platelet activation resulting in thrombocytopenia.3 Deletion of IFN-α/βR1 inhibited the increase in plasma TAT complexes, D-dimer, and PAI-1 (Figure 1F). Furthermore, the loss of IFN-α/βR1 significantly attenuated endotoxemia-induced thrombocytopenia (Figure 1G). Deletion of IFN-α/βR1 significantly increased survival in mice with endotoxic shock (Figure 1H). However, deletion of IFN-α/βR1 failed to affect FeCl3-induced thrombosis (Figure 1I). Taken together, these findings indicate that type 1 IFN signaling mediates LPS-induced activation of coagulation cascades.
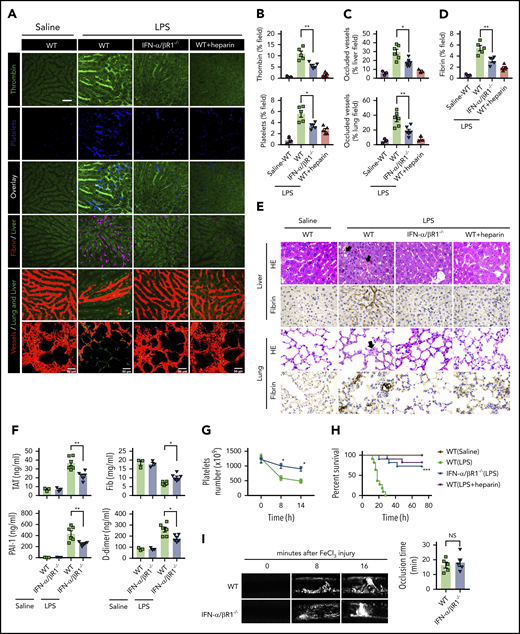
Type 1 IFN signaling mediates the activation of coagulation cascades in endotoxemia. (A-D) WT and IFN-α/βR1 KO mice were administered LPS intraperitoneally (4 mg/kg) for 6 hours. Heparin (200 IU/kg) was injected subcutaneously 30 minutes before LPS injection. (A) Representative SD-IVM images of thrombin generation (green), platelet aggregation (blue), fibrin deposition (dark red), and albumin (red) within the liver microvasculature or representative multiphoton microscopy images of albumin (red) within lung microvasculature. AF647-albumin (red) was represented as a contrast material to identify perfused vessels; the occluded vessels exhibited weak fluorescent signals. Quantitative analysis was conducted of thrombin and platelets (B), occluded vessels (C), and fibrin deposition (D) within the liver microcirculation by using ImageJ software. (E-H) Mice were primed with 0.4 mg/kg of LPS for 7 hours and then challenged with 10 mg/kg of LPS for 8 hours. (E) Representative images of hematoxylin and eosin and immunohistochemical staining of fibrin in livers and lungs of WT mice vs IFN-α/βR1 KO mice (400×). The black arrow indicates thrombus in liver and lung capillaries from WT mice challenged with LPS. (F) Plasma levels of TAT complexes, PAI-1, fibrinogen (Fib), and D-dimer were detected in WT mice vs IFN-α/βR1 KO mice. (G) Time course of thrombocytopenia in WT mice vs IFN-α/βR1 KO mice after administration of 10 mg/kg of LPS treated at time 0, 8, and 14 hours. (H) Kaplan-Meier survival plots for WT mice vs IFN-α/βR1 KO mice treated with saline or LPS or LPS plus heparin (n = 11 mice per group). (I) Representative images of FeCl3-induced mesenteric arteriole thrombosis in WT mice and IFN-α/βR1 KO mice (left). Occlusion time of the mesenteric arteriole was analyzed (right). All data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 3 to 11 mice per group. Scale bar, 50 μm. NS, not significant.
Type 1 IFN signaling mediates the activation of coagulation cascades in endotoxemia. (A-D) WT and IFN-α/βR1 KO mice were administered LPS intraperitoneally (4 mg/kg) for 6 hours. Heparin (200 IU/kg) was injected subcutaneously 30 minutes before LPS injection. (A) Representative SD-IVM images of thrombin generation (green), platelet aggregation (blue), fibrin deposition (dark red), and albumin (red) within the liver microvasculature or representative multiphoton microscopy images of albumin (red) within lung microvasculature. AF647-albumin (red) was represented as a contrast material to identify perfused vessels; the occluded vessels exhibited weak fluorescent signals. Quantitative analysis was conducted of thrombin and platelets (B), occluded vessels (C), and fibrin deposition (D) within the liver microcirculation by using ImageJ software. (E-H) Mice were primed with 0.4 mg/kg of LPS for 7 hours and then challenged with 10 mg/kg of LPS for 8 hours. (E) Representative images of hematoxylin and eosin and immunohistochemical staining of fibrin in livers and lungs of WT mice vs IFN-α/βR1 KO mice (400×). The black arrow indicates thrombus in liver and lung capillaries from WT mice challenged with LPS. (F) Plasma levels of TAT complexes, PAI-1, fibrinogen (Fib), and D-dimer were detected in WT mice vs IFN-α/βR1 KO mice. (G) Time course of thrombocytopenia in WT mice vs IFN-α/βR1 KO mice after administration of 10 mg/kg of LPS treated at time 0, 8, and 14 hours. (H) Kaplan-Meier survival plots for WT mice vs IFN-α/βR1 KO mice treated with saline or LPS or LPS plus heparin (n = 11 mice per group). (I) Representative images of FeCl3-induced mesenteric arteriole thrombosis in WT mice and IFN-α/βR1 KO mice (left). Occlusion time of the mesenteric arteriole was analyzed (right). All data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 3 to 11 mice per group. Scale bar, 50 μm. NS, not significant.
TRIF is critical for the activation of coagulation cascades in endotoxemia
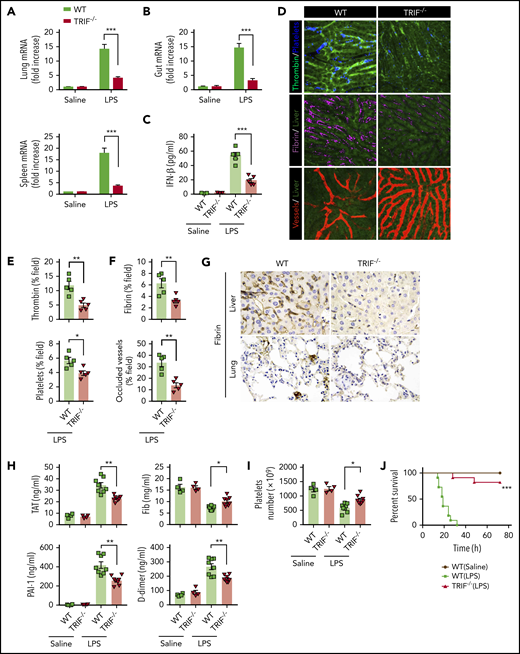
TRIF, an adaptor molecule downstream of TLR4, is required for LPS-induced expression of type 1 IFNs.20 To further confirm that type 1 IFN signaling is essential for LPS-induced activation of coagulation cascades, TRIF KO mice and their WT controls were subjected to endotoxemia. As expected, the deletion of TRIF blocked the expression of type 1 IFNs in the lungs, spleens, and guts (Figure 2A-B) and inhibited type 1 IFN secretion (Figure 2C). Deletion of TRIF markedly inhibited the intravascular thrombin generation, the aggregation of platelets, fibrin deposition, and microvessel occlusion during endotoxemia (Figure 2D-F). Fibrin deposition in the livers and lungs was further revealed by immunohistochemical staining in TRIF-deficient and WT endotoxemia mice (Figure 2G). Furthermore, loss of TRIF inhibited the decrease of platelets and the increase of plasma TAT complexes, D-dimer, and PAI-1 in endotoxemia (Figure 2H-I). Moreover, deletion of TRIF significantly promoted survival during lethal endotoxemia (Figure 2J).
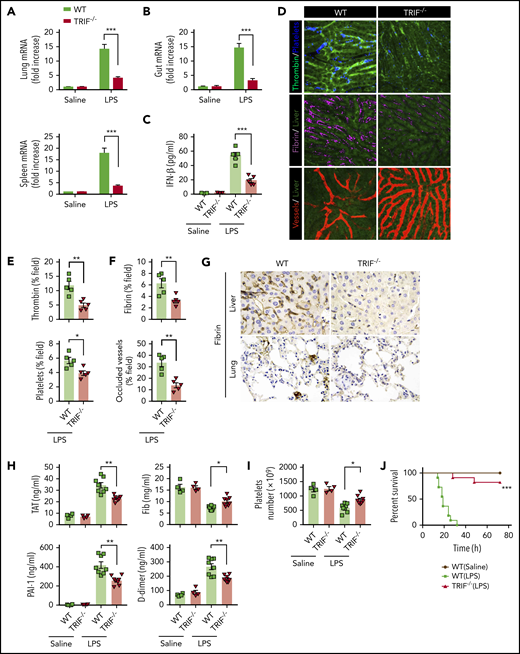
TRIF is critical for the activation of coagulation cascades in endotoxemia. (A-B) IFN-β messenger RNA (mRNA) expression in lungs, spleens (A), and guts (B) from WT mice vs TRIF KO mice as detected by quantitative polymerase chain reaction after LPS challenge for 2 hours. (C) Plasma levels of IFN-β detected by enzyme-linked immunosorbent assay in WT mice vs TRIF KO mice after LPS treatment (0.4 mg/kg of LPS for 7 hours + 10 mg/kg of LPS for 8 hours). (D) Representative SD-IVM images of thrombin (green), platelet adhesion (blue), fibrin (dark red), and albumin (red) within the liver microvasculature in endotoxemic WT and TRIF KO mice (4 mg/kg of LPS for 6 hours). (E-F) Quantitative analysis of thrombin, platelets, and fibrin probe fluorescence intensity and occluded vessels within the liver microcirculation by using ImageJ software. (G-I) WT and TRIF KO mice were injected with 0.4 mg/kg of LPS for 7 hours followed by 10 mg/kg of LPS for 8 hours. (G) Representative images of immunohistochemical staining of fibrin in livers and lungs are shown (×400). (H) Plasma levels of TAT complexes, PAI-1, fibrinogen (Fib), and D-dimer were measured. (I) Platelet counts were detected. (J) Kaplan-Meier survival plots for WT mice vs TRIF KO mice (n = 11 mice per group). Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 4 to 11 mice per group. Scale bar, 50 μm.
TRIF is critical for the activation of coagulation cascades in endotoxemia. (A-B) IFN-β messenger RNA (mRNA) expression in lungs, spleens (A), and guts (B) from WT mice vs TRIF KO mice as detected by quantitative polymerase chain reaction after LPS challenge for 2 hours. (C) Plasma levels of IFN-β detected by enzyme-linked immunosorbent assay in WT mice vs TRIF KO mice after LPS treatment (0.4 mg/kg of LPS for 7 hours + 10 mg/kg of LPS for 8 hours). (D) Representative SD-IVM images of thrombin (green), platelet adhesion (blue), fibrin (dark red), and albumin (red) within the liver microvasculature in endotoxemic WT and TRIF KO mice (4 mg/kg of LPS for 6 hours). (E-F) Quantitative analysis of thrombin, platelets, and fibrin probe fluorescence intensity and occluded vessels within the liver microcirculation by using ImageJ software. (G-I) WT and TRIF KO mice were injected with 0.4 mg/kg of LPS for 7 hours followed by 10 mg/kg of LPS for 8 hours. (G) Representative images of immunohistochemical staining of fibrin in livers and lungs are shown (×400). (H) Plasma levels of TAT complexes, PAI-1, fibrinogen (Fib), and D-dimer were measured. (I) Platelet counts were detected. (J) Kaplan-Meier survival plots for WT mice vs TRIF KO mice (n = 11 mice per group). Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 4 to 11 mice per group. Scale bar, 50 μm.
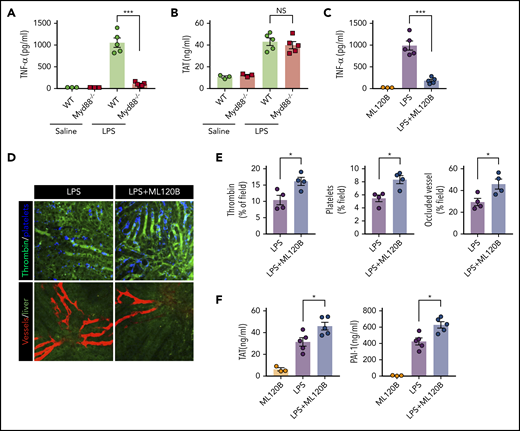
MyD88, another adaptor molecule downstream of TLR4, is important for LPS-induced production of proinflammatory cytokines.39 To investigate the role of MyD88 in LPS-induced coagulation, MyD88-deficient mice and their WT controls were challenged with LPS. Deletion of MyD88 completely blocked tumor necrosis factor (TNF) release during endotoxemia (Figure 3A). By contrast, MyD88 deficiency only slightly attenuated LPS-induced coagulation (Figure 3B). In addition to inducing the production of type 1 IFNs, the TRIF signaling is able to activate the NF-κB pathway.40 To determine the role of NF-kB activation in LPS-induced coagulation, we pharmacologically inhibited the activation of IκB kinase β (IKKβ), which is essential for LPS-induced NF-κB activation. Administration of the IKKβ inhibitor ML120B blocked TNF release but enhanced thrombin generation, platelet aggregation, and microvasculature occlusion during endotoxemia (Figure 3C-E). Furthermore, administration of ML120B promoted TAT complexes and PAI-1 increases in endotoxemia (Figure 3F). These findings are in line with a previous report showing that pharmacologic inhibition of IKKβ or genetic deletion of IKKβ in myeloid cells promotes LPS-induced lethality.41 Taken together, these findings further support the notion that type 1 IFN signaling mediates LPS-induced activation of coagulation cascades.
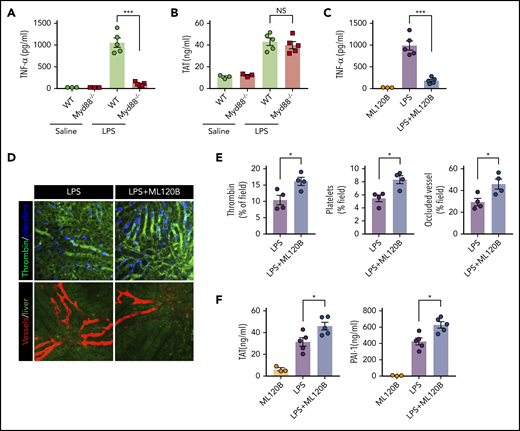
Inhibition of NF-kB activation enhanced LPS-induced coagulation. (A) Plasma concentration of TNF-α was measured at 1 hour after Myd88-deficient and WT mice were injected with 0.4 mg/kg of LPS. (B) Plasma concentration of TAT was measured in Myd88-deficient and WT mice primed with 0.4 mg/kg of LPS and then challenged with 10 mg/kg of LPS for 8 hours. (C-F) WT mice were treated with or without the IKKβ inhibitor ML120B (400 mg/kg by oral gavage twice daily for 4 days). Plasma concentration of TNF-α was measured at 1 hour after 0.4 mg/kg of LPS was injected into mice pretreated with or without ML120B (C). Representative SD-IVM images of thrombin (green), platelet adhesion (blue), and albumin (red) within the liver microvasculature in mice pretreated with or without ML120B, and then challenged with 4 mg/kg of LPS for 6 hours (D). Quantitative analysis of thrombin, platelets, and occluded vessels within the liver microcirculation by using ImageJ software (E). Plasma concentration of TAT complexes and PAI-1 were detected in mice pretreated with or without ML120B, and primed with 0.4 mg/kg of LPS for 7 hours followed by10 mg/kg of LPS for 8 hours (F). *P < .05; ***P < .001. NS, not significant.
Inhibition of NF-kB activation enhanced LPS-induced coagulation. (A) Plasma concentration of TNF-α was measured at 1 hour after Myd88-deficient and WT mice were injected with 0.4 mg/kg of LPS. (B) Plasma concentration of TAT was measured in Myd88-deficient and WT mice primed with 0.4 mg/kg of LPS and then challenged with 10 mg/kg of LPS for 8 hours. (C-F) WT mice were treated with or without the IKKβ inhibitor ML120B (400 mg/kg by oral gavage twice daily for 4 days). Plasma concentration of TNF-α was measured at 1 hour after 0.4 mg/kg of LPS was injected into mice pretreated with or without ML120B (C). Representative SD-IVM images of thrombin (green), platelet adhesion (blue), and albumin (red) within the liver microvasculature in mice pretreated with or without ML120B, and then challenged with 4 mg/kg of LPS for 6 hours (D). Quantitative analysis of thrombin, platelets, and occluded vessels within the liver microcirculation by using ImageJ software (E). Plasma concentration of TAT complexes and PAI-1 were detected in mice pretreated with or without ML120B, and primed with 0.4 mg/kg of LPS for 7 hours followed by10 mg/kg of LPS for 8 hours (F). *P < .05; ***P < .001. NS, not significant.
Type 1 IFN signaling mediates the activation of coagulation cascades in bacterial sepsis
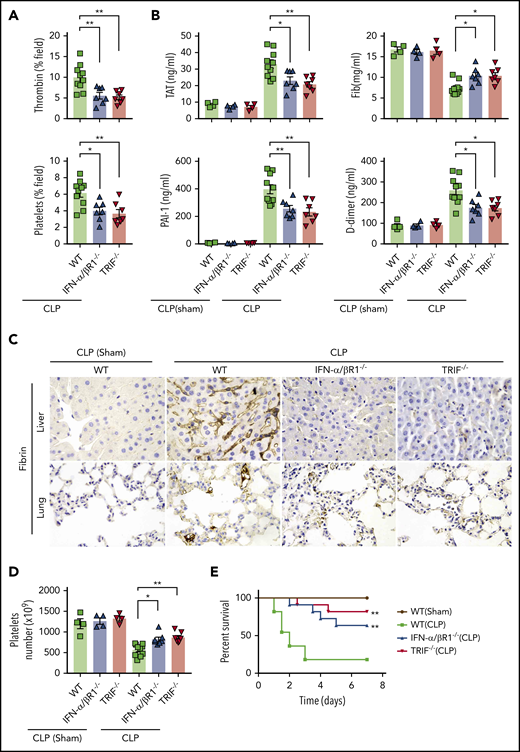
To determine whether type 1 IFN signaling mediates coagulopathy in bacterial sepsis, IFN-α/βR1 KO mice, TRIF KO mice, and their WT controls were subjected to CLP, a clinically relevant murine model of polymicrobial sepsis.8 Notably, CLP induced robust thrombin generation and platelet aggregation within the liver microvasculature of WT mice but not IFN-α/βR1 KO or TRIF KO mice (Figure 4A). Fibrin deposition in the livers and lungs were revealed by immunohistochemical staining in CLP-induced sepsis in mice (Figure 4C). Consistently, the loss of IFN-α/βR1 or TRIF prevented the decrease of platelets and the increase of plasma TAT complexes, D-dimer, and PAI-1 (Figure 4B,D). Furthermore, IFN-α/βR1 deficiency or TRIF deficiency slightly decreased the bacterial loads in the livers, lungs, and spleens (supplemental Figure 1) and markedly improved survival in experimental sepsis (Figure 4E). Thus, type 1 IFN signaling mediates the activation of coagulation cascades in bacterial sepsis.
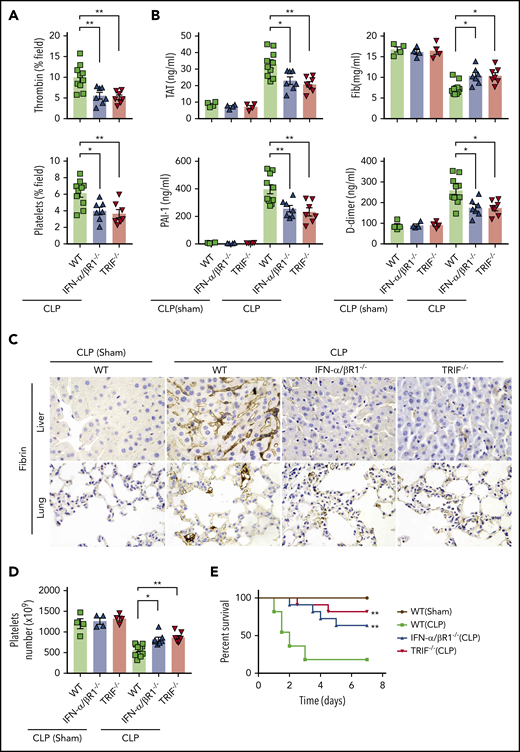
Type 1 IFN signaling mediates the activation of coagulation cascades in bacterial sepsis. (A-D) WT, IFN-α/βR1 KO, and TRIF KO mice subjected to either CLP or sham operation for 16 hours (WT group is the mixture of WT littermates of IFN-α/βR1 KO and TRIF KO mice). Thrombin and platelet fluorescence intensity in the liver microcirculation was quantified by using ImageJ software (A). Plasma levels of TAT complexes, PAI-1, fibrinogen (Fib), and D-dimer were measured (B). Representative images of immunohistochemical staining of fibrin in livers and lungs (×400) (C). Platelet counts in 16 hours after CLP- or sham-treated mice of indicated genotypes (D). (E) Kaplan-Meier survival plots for mice subjected to either CLP or sham operation (N = 11 mice per group). Data are shown as mean ± standard error of the mean. *P < .05; **P < .01. N = 4 to 11 mice per group.
Type 1 IFN signaling mediates the activation of coagulation cascades in bacterial sepsis. (A-D) WT, IFN-α/βR1 KO, and TRIF KO mice subjected to either CLP or sham operation for 16 hours (WT group is the mixture of WT littermates of IFN-α/βR1 KO and TRIF KO mice). Thrombin and platelet fluorescence intensity in the liver microcirculation was quantified by using ImageJ software (A). Plasma levels of TAT complexes, PAI-1, fibrinogen (Fib), and D-dimer were measured (B). Representative images of immunohistochemical staining of fibrin in livers and lungs (×400) (C). Platelet counts in 16 hours after CLP- or sham-treated mice of indicated genotypes (D). (E) Kaplan-Meier survival plots for mice subjected to either CLP or sham operation (N = 11 mice per group). Data are shown as mean ± standard error of the mean. *P < .05; **P < .01. N = 4 to 11 mice per group.
Type 1 IFN signaling mediates the activation of coagulation cascades by amplifying the release of HMGB1 into the bloodstream
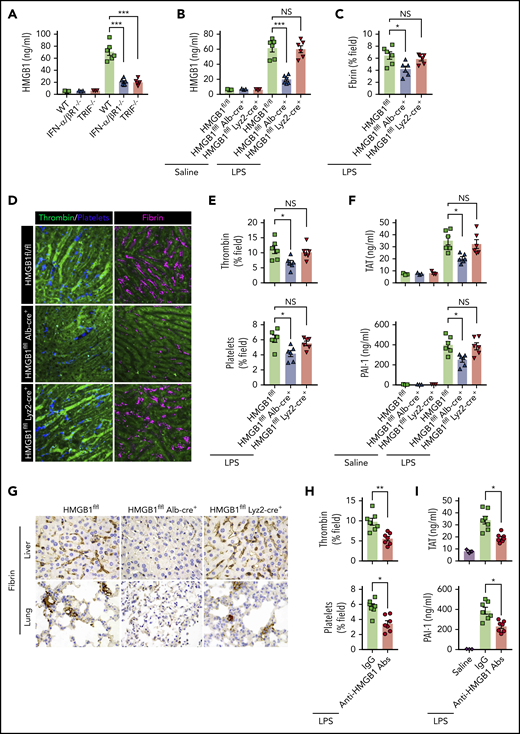
We next investigated the mechanisms by which type 1 IFN signaling mediates LPS-induced activation of coagulation cascades. HMGB1 is an evolutionarily conserved protein that is expressed virtually in all types of cells in mammals.18 Bacterial sepsis or endotoxemia leads to systemic accumulation of HMGB1 in the circulation.26,38 Neutralizing circulating HMGB1 by monoclonal antibodies significantly promotes survival in bacterial sepsis.42 We previously found that the type 1 IFN signaling mediates HMGB1 hyperacetylation at its nuclear location sequences, culminating in HMGB1 accumulation in the cytoplasm and subsequent release into the extracellular space.43 In line with a previous study,44 deletion of TRIF or IFN-α/βR1 blocked HMGB1 release in endotoxemia (Figure 5A). By contrast, deletion of MyD88 failed to inhibit LPS-induced HMGB1 release (supplemental Figure 2). We also observed that plasma levels of TAT were correlated with the concentrations of circulating HMGB1 but not with the plasma levels of TNF in both endotoxemia and bacterial sepsis (supplemental Figure 3).
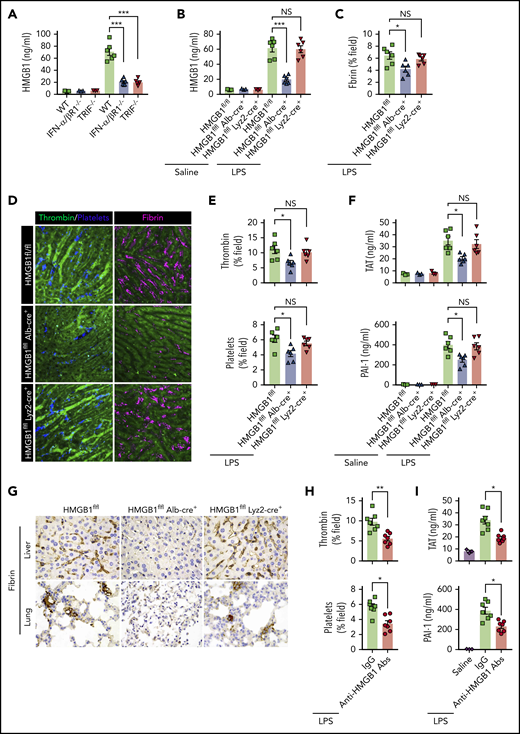
Type 1 IFN signaling mediates the activation of coagulation cascades by amplifying the release of HMGB1 into the bloodstream. (A-B) Plasma HMGB1 concentration was detected in mice of indicated genotypes primed with 0.4 mg/kg of LPS for 7 hours and then challenged with 10 mg/kg of LPS for 8 hours. (C) Quantitative analysis of fibrin in the liver microcirculation in SD-IVM images. (D) Representative SD-IVM images of thrombin (green), platelets (blue), and fibrin (dark red, AF594) in the liver microvasculature. (E) Quantitative analysis of thrombin generation and platelet activation in the liver microvasculature. (F-G) Mice of indicated genotypes were primed with 0.4 mg/kg of LPS for 7 hours and then challenged with 10 mg/kg of LPS for 8 hours. Plasma TAT and PAI-1 concentrations were detected (F). Representative images of immunohistochemical staining of fibrin in livers and lungs are shown (G) (×400). (H-I) WT mice were injected with or without monoclonal HMGB1 neutralizing antibody (2G7, 160 μg/mouse) or the isotype control IgG (160 μg/mouse) 30 minutes before administration of 10 mg/kg of LPS (TAT and PAI-1) or 4 mg/kg of LPS (SD-IVM). Quantitative analysis of thrombin generation and platelet activation within the liver microvasculature (H) and plasma levels of TAT complexes and PAI-1 (I) are shown. Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 3 to 7 mice per group. Scale bar, 50 μm. NS, no significant.
Type 1 IFN signaling mediates the activation of coagulation cascades by amplifying the release of HMGB1 into the bloodstream. (A-B) Plasma HMGB1 concentration was detected in mice of indicated genotypes primed with 0.4 mg/kg of LPS for 7 hours and then challenged with 10 mg/kg of LPS for 8 hours. (C) Quantitative analysis of fibrin in the liver microcirculation in SD-IVM images. (D) Representative SD-IVM images of thrombin (green), platelets (blue), and fibrin (dark red, AF594) in the liver microvasculature. (E) Quantitative analysis of thrombin generation and platelet activation in the liver microvasculature. (F-G) Mice of indicated genotypes were primed with 0.4 mg/kg of LPS for 7 hours and then challenged with 10 mg/kg of LPS for 8 hours. Plasma TAT and PAI-1 concentrations were detected (F). Representative images of immunohistochemical staining of fibrin in livers and lungs are shown (G) (×400). (H-I) WT mice were injected with or without monoclonal HMGB1 neutralizing antibody (2G7, 160 μg/mouse) or the isotype control IgG (160 μg/mouse) 30 minutes before administration of 10 mg/kg of LPS (TAT and PAI-1) or 4 mg/kg of LPS (SD-IVM). Quantitative analysis of thrombin generation and platelet activation within the liver microvasculature (H) and plasma levels of TAT complexes and PAI-1 (I) are shown. Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 3 to 7 mice per group. Scale bar, 50 μm. NS, no significant.
Together with the finding that plasma HMGB1 levels correlate with DIC scores in patients with sepsis,45 we postulate that type 1 IFN signaling might mediate LPS-induced activation of coagulation cascades through HMGB1 release into the circulation. Because both myeloid cells and hepatocytes have been shown to actively and passively release HMGB1 during inflammation or hypoxia,38,46 we generated mice with selective HMGB1 deletion in either myeloid cells (Hmgb1f/f Lyz2-cre+) or hepatocytes (Hmgb1f/f Alb-cre+). Although global deletion of Hmgb1 leads to early postnatal death, these transgenic mice displayed no detectable defects under physiological conditions.21 Consistent with our previous observations, we found that hepatocytes, but not myeloid cells, were the major source of the increased plasma HMGB1 concentrations during endotoxemia (Figure 5B). Notably, deletion of hepatocyte, but not myeloid, HMGB1 significantly attenuated fibrin deposition in the livers during endotoxemia (Figure 5C-D). Loss of hepatocyte HMGB1 significantly inhibited the generation of intravascular thrombin and the aggregation of platelets during endotoxemia (Figure 5D-E). HMGB1 deficiency in hepatocytes reduced plasma TAT complexes and PAI-1 in endotoxemia (Figure 5F). Fibrin deposition in the livers and lungs was detected by immunohistochemical staining (Figure 5G). Previous studies have reported that platelets are the major source of HMGB1 within thrombi and platelet-derived HMGB1 as a critical mediator for injury-induced thrombosis in vivo.47 However, using 2 kinds of transgenic mouse models that special ablation of HMGB1 in platelets (HMGB1fl/fl,Pf4-cre+ mice)22 or HMGB1 deleted in hematopoietic cells including progenitors (HMGB1fl/fl,Vav-cre+ mice).24 We found that platelet-derived HMGB1 was not the main source of HMGB1, and HMGB1 deficiency in platelet or hematopoietic cells could not prevent the increase of plasma TAT complexes and PAI-1 in endotoxemia and sepsis (supplemental Figure 4). Importantly, administration of anti-HMGB1 monoclonal antibodies significantly attenuated LPS-induced coagulopathy in a manner similar to that of deletion of HMGB1 (Figure 5H-I). Together, our data establish that type 1 IFN signaling mediates the activation of coagulation cascades, at least in part, through regulating HMGB1 release into the bloodstream.
Type 1 IFN signaling and HMGB1 mediate TF-dependent coagulopathy in endotoxemia through PS exposure
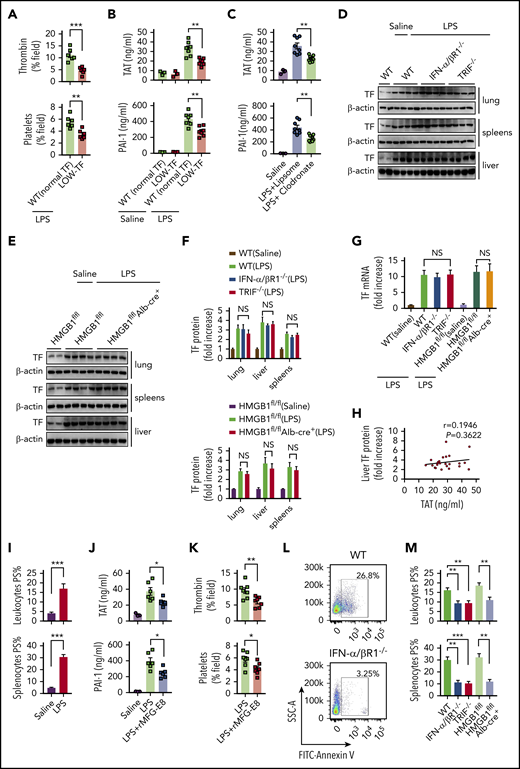
We next investigated the mechanisms by which type 1 IFNs and HMGB1 mediate LPS-induced coagulopathy. Similar to previous results, mice expressing low levels of TF have markedly reduced activation of coagulation in endotoxemia (Figure 6A-B), indicating a critical role of TF in the development of LPS-induced DIC. TF expressed in myeloid cells (eg, macrophages) plays a critical role in the development of DIC.12 In line with this finding, depletion of macrophages using clodronate liposomes30,31 significantly attenuated thrombin generation and platelet aggregation (supplemental Figure 5) and decreased plasma concentrations of TAT complexes and PAI-1 in endotoxemia (Figure 6C). Because a previous study showed that recombinant HMGB1 is able to stimulate the expression of TF in macrophages and endothelial cells,48 we next tested whether HMGB1 mediates LPS-induced coagulopathy through upregulation of TF expression. However, deletion of IFN-α/βR1, TRIF, or hepatocyte HMGB1 failed to affect the TF expression in the lungs, livers, and spleens (Figure 6D-G). Levels of TF expression did not correlate with plasma levels of TAT (Figure 6H). These observations prompted us to test whether the type 1 IFN/HMGB1 pathway promotes TF activity in endotoxemia at the posttranslational level.
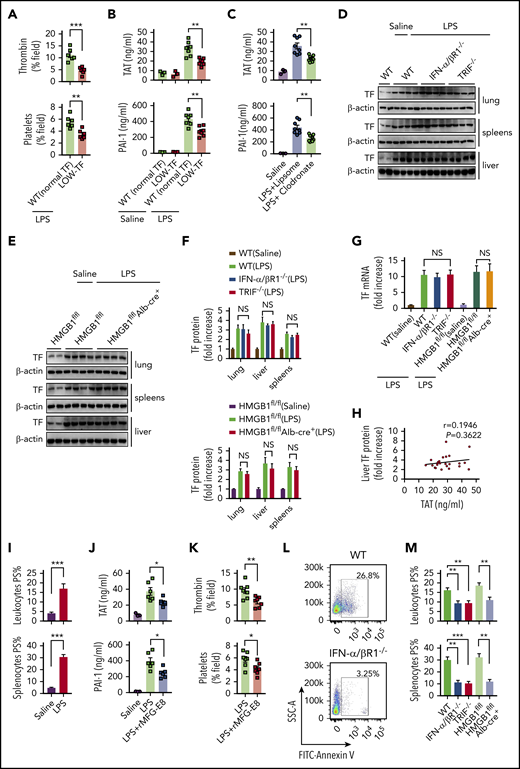
Type 1 IFN signaling and HMGB1 mediates TF-dependent coagulopathy in endotoxemia through PS exposure. (A) Quantitative analysis SD-IVM images of thrombin and platelet fluorescence intensity within the liver microcirculation from endotoxemic mice expressing normal or low levels of TF (4 mg/kg of LPS for 6 hours). (B) Plasma levels of TAT complexes and PAI-1 in saline- and LPS-challenged normal or low levels of TF mice (0.4 mg/kg of LPS for 7 hours + 10 mg/kg of LPS for 8 hours). (C) Mice were injected with liposome-clodronate and liposome–phosphate-buffered saline before LPS injection (0.4 mg/kg of LPS for 7 hours + 10 mg/kg of LPS for 8 hours). Plasma concentrations of TAT and PAI-1 were measured. (D-E) TF protein detected by western blot in the lungs, livers, and spleens from WT, IFN-α/βR1 KO, TRIF KO, Hmgb1fl/fl, and Hmgb1fl/fl Alb-cre+ mice that were challenged with LPS. (F-G) Quantitative analysis of TF protein and messenger RNA (mRNA) (liver) expression. Values are given as fold increase over unstimulated controls. (H) The correction between TF protein in liver detected by using enzyme-linked immunosorbent assay and plasma concentrations of TAT. (I) Quantitative analysis of PS exposure in peripheral leukocytes and splenocytes from endotoxemic WT mice by using FlowJo software. (J-K) WT mice were injected with or without recombinant MFG-E8 (rMFG-E8; 160 μg/kg) 2 hours before administration of 10 mg/kg of LPS (TAT and PAI-1) or 4 mg/kg of LPS (SD-IVM). Plasma levels of TAT complexes and PAI-1 (J) and quantitative analysis of thrombin and platelet fluorescence intensity within the liver microcirculation (K). (L-M) Flow cytometric analysis of PS exposure labeled by fluorescein isothiocyanate (FITC)–AnnexinV in peripheral leukocytes and splenocytes from mice of indicated genotypes. Representative images of PS exposure in splenocytes (L). Quantitative analysis of PS exposure in peripheral leukocytes and splenocytes (M). Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 3 to 12 mice per group. NS, not significant.
Type 1 IFN signaling and HMGB1 mediates TF-dependent coagulopathy in endotoxemia through PS exposure. (A) Quantitative analysis SD-IVM images of thrombin and platelet fluorescence intensity within the liver microcirculation from endotoxemic mice expressing normal or low levels of TF (4 mg/kg of LPS for 6 hours). (B) Plasma levels of TAT complexes and PAI-1 in saline- and LPS-challenged normal or low levels of TF mice (0.4 mg/kg of LPS for 7 hours + 10 mg/kg of LPS for 8 hours). (C) Mice were injected with liposome-clodronate and liposome–phosphate-buffered saline before LPS injection (0.4 mg/kg of LPS for 7 hours + 10 mg/kg of LPS for 8 hours). Plasma concentrations of TAT and PAI-1 were measured. (D-E) TF protein detected by western blot in the lungs, livers, and spleens from WT, IFN-α/βR1 KO, TRIF KO, Hmgb1fl/fl, and Hmgb1fl/fl Alb-cre+ mice that were challenged with LPS. (F-G) Quantitative analysis of TF protein and messenger RNA (mRNA) (liver) expression. Values are given as fold increase over unstimulated controls. (H) The correction between TF protein in liver detected by using enzyme-linked immunosorbent assay and plasma concentrations of TAT. (I) Quantitative analysis of PS exposure in peripheral leukocytes and splenocytes from endotoxemic WT mice by using FlowJo software. (J-K) WT mice were injected with or without recombinant MFG-E8 (rMFG-E8; 160 μg/kg) 2 hours before administration of 10 mg/kg of LPS (TAT and PAI-1) or 4 mg/kg of LPS (SD-IVM). Plasma levels of TAT complexes and PAI-1 (J) and quantitative analysis of thrombin and platelet fluorescence intensity within the liver microcirculation (K). (L-M) Flow cytometric analysis of PS exposure labeled by fluorescein isothiocyanate (FITC)–AnnexinV in peripheral leukocytes and splenocytes from mice of indicated genotypes. Representative images of PS exposure in splenocytes (L). Quantitative analysis of PS exposure in peripheral leukocytes and splenocytes (M). Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. N = 3 to 12 mice per group. NS, not significant.
PS increases TF activity and enhances the assembly cofactor-protease complexes of the coagulation cascade.11,14 Under physiological conditions, most PS are localized to the inner layer of the cytoplasmic membrane.15 Gram-negative sepsis induces PS externalization in peripheral leukocytes,16 but the blockage of PS on the cell surface significantly attenuates bacterial sepsis–induced DIC.16,17 In agreement with previous findings,16 LPS challenge induced robust PS externalization in peripheral leukocytes and splenocytes (Figure 6I). Administration of MFG-E8, a well-known PS-binding protein,15,49 significantly attenuated endotoxin-induced coagulopathy (Figure 6J-K). Notably, the deletion of IFN-α/βR1, TRIF, or hepatocyte HMGB1 uniformly decreased endotoxin-induced PS externalization in peripheral leukocytes and splenocytes (Figure 6L-M). Taken together, these data show that type 1 IFNs and HMGB1 mediate TF-dependent coagulopathy in endotoxemia, at least in part, through PS exposure.
Extracellular HMGB1 induces PS exposure and TF activation through caspase-11
We next investigated the mechanisms by which extracellular HMGB1 mediates PS exposure and TF-dependent coagulopathy. Recent advances have shown that caspase-11 and its substrate gasdermin D (GSDMD) trigger TF-dependent activation of coagulation cascades in endotoxemia.50 Together with our recent finding that HMGB1 mediates caspase-11–dependent lethality in sepsis by delivery of LPS into the cytoplasm,21 we reasoned that extracellular HMGB1 might induce PS externalization and TF activation in the presence of LPS through caspase-11. As shown by confocal microscopy, recombinant HMGB1 or ultrapure LPS alone failed to induce detectable PS externalization in cultured mouse peritoneal macrophages (Figure 7A-B). However, the addition of both HMGB1 and ultrapure LPS induced robust PS exposure in WT but not caspase-11–deficient mouse peritoneal macrophages. Accordingly, HMGB1 increased the TF procoagulant activity in WT but not in caspase-11–deficient mouse peritoneal macrophages in a time-dependent manner (Figure 7C-D). HMGB1 failed to induce PS externalization in GSDMD KO mouse peritoneal macrophages in the presence of LPS (Figure 7E).
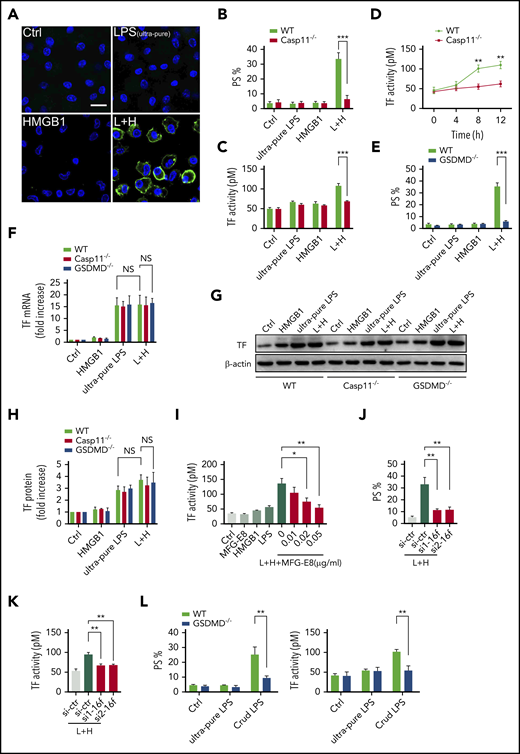
Extracellular HMGB1 induces PS exposure and TF activation through caspase-11. (A-C, E, G-H) WT, caspase-11 KO, and GSDMD KO macrophages stimulated with ultrapure LPS (L, 1 μg/mL) or HMGB1 (H, 400 ng/mL) or luteinizing hormone (LH) for 10 hours. Representative confocal images of PS exposure labeled by Annexin V–fluorescein isothiocyanate are shown (A), PS exposure was quantified by using ImageJ software for the area percentage of cells that are positive (B and E), and TF activity was detected (C). (D) TF activity was measured at different time points after WT and caspase-11 KO macrophages were treated with ultrapure LPS plus HMGB1. (F) WT, caspase-11 KO, and GSDMD KO macrophages stimulated with ultrapure LPS (L, 1 μg/mL) or HMGB1 (H, 400 ng/mL), or LH for 2 hours. TF messenger RNA (mRNA) expression was detected by using quantitative polymerase chain reaction. Values are given as fold increase over unstimulated controls. (G) TF protein expression was detected by using western blot. (H) Quantitative analysis of TF protein expression. (I) TF activity was measured in HCV macrophages stimulated with HMGB1 plus ultrapure LPS with or without MFG-E8. PS exposure (J) and TF activity (K) were detected in LH-treated HCV macrophages transfected with control small interfering RNA (siRNA) or TMEM16F-specific siRNA. si1 and si2 represent 2 sequences of Tmem16f-specific siRNAs. (L) WT and GSDMD KO macrophages stimulated with ultrapure LPS (1 μg/mL) or crude LPS (1 μg/mL) for 10 hours. PS exposure was quantified (left) and TF activity was detected (right). HCV mice were used for the aforementioned TF activity tests. Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. NS, not significant.
Extracellular HMGB1 induces PS exposure and TF activation through caspase-11. (A-C, E, G-H) WT, caspase-11 KO, and GSDMD KO macrophages stimulated with ultrapure LPS (L, 1 μg/mL) or HMGB1 (H, 400 ng/mL) or luteinizing hormone (LH) for 10 hours. Representative confocal images of PS exposure labeled by Annexin V–fluorescein isothiocyanate are shown (A), PS exposure was quantified by using ImageJ software for the area percentage of cells that are positive (B and E), and TF activity was detected (C). (D) TF activity was measured at different time points after WT and caspase-11 KO macrophages were treated with ultrapure LPS plus HMGB1. (F) WT, caspase-11 KO, and GSDMD KO macrophages stimulated with ultrapure LPS (L, 1 μg/mL) or HMGB1 (H, 400 ng/mL), or LH for 2 hours. TF messenger RNA (mRNA) expression was detected by using quantitative polymerase chain reaction. Values are given as fold increase over unstimulated controls. (G) TF protein expression was detected by using western blot. (H) Quantitative analysis of TF protein expression. (I) TF activity was measured in HCV macrophages stimulated with HMGB1 plus ultrapure LPS with or without MFG-E8. PS exposure (J) and TF activity (K) were detected in LH-treated HCV macrophages transfected with control small interfering RNA (siRNA) or TMEM16F-specific siRNA. si1 and si2 represent 2 sequences of Tmem16f-specific siRNAs. (L) WT and GSDMD KO macrophages stimulated with ultrapure LPS (1 μg/mL) or crude LPS (1 μg/mL) for 10 hours. PS exposure was quantified (left) and TF activity was detected (right). HCV mice were used for the aforementioned TF activity tests. Data are shown as mean ± standard error of the mean. *P < .05; **P < .01; ***P < .001. NS, not significant.
Consistent with the results of in vivo experiments, extracellular HMGB1 did not affect the expression of TF in the presence of LPS (Figure 7F-H). Neutralizing PS using MFG-E8 markedly decreased the cell surface TF activity without altering the expression of TF (Figure 7I; supplemental Figure 6A). Furthermore, reducing the expression of TMEM16F, a phospholipid scramblase required for PS exposure upon GSDMD activation,51 significantly inhibited PS exposure and TF activation in HMGB1 plus LPS-stimulated macrophages without affecting TF expression (Figure 7J-K; supplemental Figure 6B). Unlike ultrapure LPS, crude LPS is known to activate inflammasomes.52 Accordingly, crude LPS but not ultrapure LPS induced PS exposure and TF activation in a GSDMD-dependent manner (Figure 7L). Furthermore, deletion of caspase-11 or administration of heparin significantly reduced plasma levels of TAT and PAI-1 in endotoxemia (supplemental Figure 7). These findings indicate that extracellular HMGB1 induces PS exposure and TF activation in the presence of LPS through caspase-11–dependent and GSDMD-dependent mechanisms.
Discussion
Both virus and gram-negative bacteria could induce the expression and release of type 1 IFNs,18 which act through IFN-α/βR to render resistance to viral infection.18,19 However, the roles of IFNs in gram-negative bacterial infection remain largely unknown. Here we report the previously underappreciated critical role for type 1 IFNs in gram-negative bacteria-induced coagulation. To maintain hemostasis, coagulation is activated as part of the host innate immune system to restrict the widespread dissemination of pathogenic bacteria.53 In line with this notion, it has been shown that genetic deletion of TRIF, an adaptor molecule downstream of TLR4 that is required for LPS-induced expression of type 1 IFNs,20 significantly increases bacterial loads in tissues during Escherichia coli infection.54 However, when local gram-negative bacterial infection escalates into sepsis, this immune-thrombotic response becomes widespread and leads to life-threatening DIC, which promotes multiple organ failures and significantly increases mortality in patients with sepsis.2-4 In addition to viral or bacterial infection, type 1 IFN signaling is augmented in patients with systemic lupus erythematosus.55 Thus, our study might explain a clinical paradox that sepsis is often a frequent and severe complication for patients with systemic lupus erythematosus, an autoimmune disease associated with elevated type 1 IFN responses.56
Mechanistically, type 1 IFNs mediate gram-negative bacteria- or LPS-induced coagulation through triggering the release of effector molecules (eg, HMGB1) into the bloodstream. HMGB1 is a widely expressed nonhistone chromatin-binding protein that normally resides in the nucleus of many types of cells.38,43 Upon bacterial or viral infection, type 1 IFN signaling could induce robust HMGB1 hyperacetylation at the nuclear location sequences, resulting in HMGB1 translocation into the cytoplasm and subsequent release into the extracellular space.21 In the context of endotoxemia or gram-negative sepsis, extracellular HMGB1 binds to and delivers LPS into the cytoplasm of the host cell, culminating in the activation of caspase-11. The inhibition of type 1 IFN production and signaling and HMGB1 expression or activities uniformly blocks caspase-11–dependent immune responses and lethality in endotoxemia and bacterial sepsis.21,57 In line with our findings, a recent study showed that caspase-11 activation induces coagulation and death through GSDMD in endotoxemia.50 Upon cleavage by caspase-11, GSDMD oligomerizes and forms nano-pores in the cytoplasm membrane.58 This action is followed by PS exposure, which increases TF activity and coagulation cascades. Intriguingly, pharmacologic inhibition of the NF-kB pathway enhanced inhibition of the activation of IKKβ, or the downstream NF-kB pathway could markedly enhance LPS-induced caspase-1 activation.59 Because activation of caspase-1 or caspase-11 triggers coagulation through GSDMD,50 it is conceivable that inhibition of NF-kB activation enhances coagulation through promotion of GSDMD activation and PS exposure.
In addition to GSDMD-dependent PS exposure, the type 1 IFN pathway might contribute to the development of DIC through promoting thiol-disulfide exchange, NETosis, or platelet activation. The protein disulfide isomerase–mediated thiol-disulfide exchange has been proven to be critical for TF activation,14,15,17 which could be inhibited by an anti–protein disulfide isomerase antibody.60 Intriguingly, the extracellular adenosine triphosphate–induced caspase-1 activation mediates the generation of extracellular thiol pathway–dependent, procoagulant microparticles.61,62 Because caspase-1 is also capable of activating GSDMD, it is important to investigate whether the type 1 IFN/HMGB1/caspase-11 pathway also promotes TF activation through GSDMD-dependent thiol-disulfide exchange in the future. Caspase-11 and GSDMD are expressed in neutrophils, and the activation of caspase-11 results in NETosis, a lytic form of cell death that releases chromatin structures to the extracellular space.63,64 These extracellular neutrophil-derived chromatin structures, termed neutrophil extracellular traps, not only prevent the dissemination of invading microbes but also enhance thrombosis.65 Thus, it is possible that the type 1 IFN pathway and caspase-11 pathway might contribute to the development of DIC, in part, through NETosis. Recent advances show that extracellular HMGB1 promotes platelet activation, granule secretion, adhesion, and spreading in the absence of LPS.47,66 This event is mediated by HMGB1–TLR4 interaction on the cell surface of platelets and MyD88-dependent recruitment of platelet guanylyl cyclase toward the plasma membrane.47
In line with these findings, we observed that deletion of IFN-α/βR1 or hepatocyte HMGB1 reduces LPS-induced platelet aggregation in the microvasculature. Intriguingly, activated platelets are able to actively release HMGB1 into extracellular space, leading to enhanced thrombosis.47,66 Although we and others have shown that hepatocytes are the major source of circulating HMGB1,21 we further showed that platelet-released HMGB1 has not contributed to the caspase-11–dependent immune responses and the development of DIC in sepsis. Reduced endothelial Tie2 signaling promotes DIC in sepsis.67 Because HMGB1 could enable LPS to cause caspase-11–dependent pyroptosis or dysfunction of endothelial cells,21 it is also possible that the type 1 IFN/HMGB1/caspase-11 pathway might promote DIC through disruption of the endothelial Tie2 axis. Gram-positive bacteria do not activate caspase-11, and thus it is unlikely that the type 1 IFN/HMGB1 pathway mediates DIC during gram-positive bacterial infection. Instead, peptidoglycan, a component of gram-positive bacteria, induces DIC through activation of both intrinsic and extrinsic coagulation pathways.68 Taken together, this study not only identified a previously unrecognized biological function of type 1 IFNs in host antibacterial immune responses, but the results might also open a new avenue for the prevention or treatment of DIC in gram-negative bacterial sepsis.
Original data can be obtained by contacting the corresponding author (Ben Lu; e-mail: xybenlu@csu.edu.cn).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to Nigel Mackman for providing the HCV and LTF mice and anti-mouse fibrin antibodies. The authors thank Q. Xue for managing the mouse colonies and research assistance, as well as the imaging core of Shanghai Institute of Immunology for their support.
This study was supported by the National Key Scientific Project 2015CB910700 (B.L.), the National Natural Science Foundation of China (No. 81930059, B.L.; No. 81971893, Y.T.; and No. 81570117, F.C.), and Innovation-driven scientific project of CSU (B.L.). H.W. is supported by the US National Center for Complementary and Alternative Medicine (R01AT005076) and the National Institutes of Health, National Institute of General Medical Sciences (Grant R01GM063075).
Authorship
Contribution: B.L. conceived the project, designed the experiments, and wrote the manuscript; Y.T., F.C., X.X., T.R.B., H.W., and J. Wu. interpreted the data, commented, and edited the manuscript; X.Y., X.C., X.Q., Z.W., G.F., J. Wang. and H.K. performed the experiments; and X.Y. and X.C. analyzed the data and created the figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ben Lu, The 3rd Xiangya Hospital, Central South University, Changsha, 410000 People’s Republic of China; e-mail: xybenlu@csu.edu.cn.