

In this issue of Blood, report on the development of myeloid malignancy in 3 individuals with homozygous sickle cell disease (SCD).1 This represented a total of 4% (3 of 76) of their cohort transplanted for SCD from 2004 to 2018. Participants with severe SCD had 4 common features: (1) before transplant, clonal hematopoiesis of indeterminate potential (CHIP)-related mutations were detected in the blood of both individuals assessed; (2) all received nonmyeloablative, allogeneic hematopoietic cell transplant (AlloHCT) using total body irradiation (TBI) (300 to 400 cGy) and alemtuzumab-based conditioning; (3) participants received mobilized peripheral blood stem cells; (4) the myeloid malignancy occurred 2 to 5 years after a failed allograft.
Potential risk factors for cancer following allogeneic hematopoietic stem cell transplant (HCT) for SCD. GVHD, graft-versus-host disease; IST, immunosuppression therapy; RBC, red blood cells.
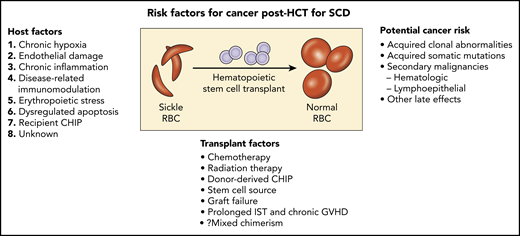
Potential risk factors for cancer following allogeneic hematopoietic stem cell transplant (HCT) for SCD. GVHD, graft-versus-host disease; IST, immunosuppression therapy; RBC, red blood cells.
In 2 large population studies, SCD patients, independent of AlloHCT, have an increased risk of developing hematology malignancies.2,3 Others have reported no increased incidence of myeloid malignancies associated with hydroxyurea therapy in SCD. Plausible underlying mechanisms for an increased risk of hematology malignancies in SCD include chronic hypoxia, endothelial damage, chronic systemic inflammation, disease-related immunomodulation, erythropoietic stress with dysregulated apoptosis, and genetic predisposition.4-6 Does the transplant regimen used play a role in this report? Baker et al showed the incidence of subsequent malignant neoplasms in patients receiving low-dose TBI regimens (200 to 450 cGy) for alloHCT was comparable to myeloablative chemotherapy (hazard ratio 1.17; 95% confidence interval 0.8 to 1.72; P = .42), but still twofold higher than a nontransplant population.7 Were there other risk factors in these patients? Two of the 3 individuals reported by Ghannam et al had evidence of progression of baseline high-risk TP53 clonal abnormalities detected by next-generation sequencing pretransplant (c.524G>A with variant allele frequency [VAF] of 72.4% in the first and c.658T>C at a VAF of 4.5% in the other). The third patient did not have a baseline blood sample analyzed.
Li et al reported 4 cases of myeloid neoplasms in individuals with SCD at a median age of 35.5 years.8 Two of these patients were treated with hydroxyurea; 2 patients were on supportive care alone, and 1 patient had undergone AlloHCT. All 4 cases demonstrated certain degrees of myelodysplasia and complex cytogenetic abnormalities with −7/7q- and/or −5/5q- or with 11q23 (KMT2A) rearrangement, similar to therapy-related myeloid neoplasm. In the report by Ghannam et al, the myeloid malignancies were seen only in patients who did not have sustained donor engraftment. Thus, the acute myeloid leukemia in these patients was due to autologous clonal proliferation of cells, rather than donor cells.
In SCD AlloHCT, simultaneous germline and somatic whole-genome sequence analysis now provides the opportunity to identify root causes of CHIP. Association of a genome-wide set of germline genetic variants has identified genetic loci associated with CHIP status, including 1 locus at TET2 that was an African ancestry–specific variant; rs144418061 in an intergenic region near TET2. Carriers of the A allele have a 2.4-fold increased risk for CHIP (P = 4.0 × 10−9). The African ancestry–specific TET2 locus risk variant disrupts the hematopoietic stem cell TET2 enhancer, whereas other variants near TET2 have been associated with myeloproliferative neoplasm.9 The incidence or progression of baseline clonal hematopoiesis in SCD with or without AlloHCT remains unknown. Patient-specific risk factors, including, age, exposure to hydroxyurea, type of transplant conditioning, stem cell source, degree of donor chimerism, and presence of donor somatic mutations, likely modulate the risk for hematologic malignancy (see figure).
Unfortunately, the life expectancy of individuals with SCD has remained relatively unchanged over the last 3 decades, currently estimated at 48 years and 54.7 years, respectively, for individuals with phenotypes HbSS/HbSβ0 thal/HbSD and HbSC/HbSβ+ thalassemia, respectively.10 Most adults with SCD also have chronic organ dysfunction, poor quality of life, and a life expectancy at least 20 years shorter than their African American counterparts living in the United States. With increasing curative options (ie, AlloHCT, myeloablative gene therapy, and gene editing), with the potential to close the disparity gap in survival, now is the time to begin to prospectively evaluate the long-term effects of these varied curative approaches for SCD. Each curative option involves the use of chemotherapy, TBI, or both with potential for causing clonal abnormalities. In addition, gene therapy and gene editing have been associated with genotoxic effects, including the potential to cause double-strand breaks at locations other than the desired genomic location. Estimation of the absolute and relative risk for treatment-related malignancy will allow a better selection of the personalized curative therapy approach that matches the cancer predisposition of each individual with SCD.
Conflict-of-interest disclosure: A.A.K. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal