

TO THE EDITOR:
Sickle cell disease (SCD) is a debilitating disease associated with extensive morbidity and early mortality. Survival in children has improved significantly during the last 4 decades as a result of newborn screening, penicillin prophylaxis, and pneumococcal vaccination.1 Although studies have not shown an increased incidence of solid tumors in adults living with SCD, 2 large population studies have revealed an increased incidence of leukemia.2,3 An association between hydroxyurea exposure in SCD and leukemia is anecdotal and unproven.4-14 Allogeneic hematopoietic cell transplantation (AlloHCT) remains the most widely available cure that can be offered to patients with SCD, with the goal of not only reversing the disease but perhaps also prolonging survival. We and others have previously reported myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML) after unsuccessful AlloHCT.8,15,16 The underlying etiology of this complication is unknown. We therefore performed detailed genomic leukemic characterization of 2 of the 3 patients who developed myeloid malignancy posttransplantation out of a total of 76 adult patients who received an AlloHCT for SCD at the National Institutes of Health Clinical Center between September 2004 and April 2018. We further sought to evaluate whether mutations present at the time of malignancy diagnosis were also detectable before transplantation.
All patients were enrolled on protocols (ClinicalTrials.gov identifiers: NCT00977691 or NCT00061658) approved by the National Heart, Lung, and Blood Institute Institutional Review Board after providing written informed consent. Next-generation sequencing (NGS), using DNA from bone marrow at the time of myeloid malignancy diagnosis, was performed clinically and confirmed using an anchored multiplex polymerase chain reaction (PCR)-based panel incorporating molecular barcode/unique molecular identifier designed to cover regions of commonly mutated genes in MDS and AML. Paired-end 150-bp sequencing used unique dual sample indices on an Illumina HiSeq 2500 (rapid run mode). Data were analyzed using Archer Analysis software (version 6.0.4). Digital droplet PCR (ddPCR) was performed for research purposes, using approximately 1 μg DNA isolated from blood, using Custom TaqMan SNP Genotyping Assays (#4351379; Thermo Fisher Scientific, Waltham, MA) on the RainDrop platform (RainDance Technologies, Lexington, MA). Data were analyzed using RainDrop Analyst II software.
All 3 patients had homozygous SCD (HbSS) and received a nonmyeloablative mobilized peripheral blood AlloHCT at the National Institutes of Health Clinical Center (Table 1).
Characteristics of patients with SCD developing therapy-related myeloid malignancy after allogeneic hematopoietic cell transplantation at the National Institutes of Health Clinical Center
| Patient ID | SCD complications | Age at AlloHCT, y | Donor type | Time from transplant to graft rejection | Time from transplant to myeloid malignancy, y | Cytogenetics at myeloid malignancy diagnosis | TP53 mutation and VAF at myeloid malignancy diagnosis |
|---|---|---|---|---|---|---|---|
| 1 | Stroke, CRI, recurrent VOC | 37 | Haplo | 73 d | 2 | Complex | c.524G>A, 72.4% in bone marrow |
| 2 | Recurrent VOC, chronic pain | 37 | HLA-matched | 6 mo | 2.5 | Complex* | c.658T>C, 4.5% in bone marrow |
| 3 | ESRD, pHTN, diastolic dysfunction | 44 | Haplo | 7 mo | 5 | 7q deletion | N/A |
| Patient ID | SCD complications | Age at AlloHCT, y | Donor type | Time from transplant to graft rejection | Time from transplant to myeloid malignancy, y | Cytogenetics at myeloid malignancy diagnosis | TP53 mutation and VAF at myeloid malignancy diagnosis |
|---|---|---|---|---|---|---|---|
| 1 | Stroke, CRI, recurrent VOC | 37 | Haplo | 73 d | 2 | Complex | c.524G>A, 72.4% in bone marrow |
| 2 | Recurrent VOC, chronic pain | 37 | HLA-matched | 6 mo | 2.5 | Complex* | c.658T>C, 4.5% in bone marrow |
| 3 | ESRD, pHTN, diastolic dysfunction | 44 | Haplo | 7 mo | 5 | 7q deletion | N/A |
A total of 76 patients received AlloHCT for HbSS at this center; myeloid malignancy was only seen within those who did not engraft
CRI, chronic renal insufficiency; ESRD, end-stage renal disease; Haplo, haploidentical donor; N/A, not available; pHTN; pulmonary hypertension.
Patient 2 had complex cytogenetics at graft rejection 2 years before formal myeloid malignancy diagnosis (no aspirate collected at a later point).
The first patient had HbSS complicated by stroke, chronic renal insufficiency with a baseline creatinine of 5 g/dL, and recurrent vaso-occlusive crises (VOC). He underwent haploidentical peripheral blood stem cell transplant (PBSCT) at 37 years of age and received alemtuzumab, 400 cGy total-body irradiation, and 100 mg/kg posttransplant cyclophosphamide in divided doses. The patient initially engrafted then rejected his graft at 73 days posttransplant. Two years posttransplant, he presented with severe neutropenia. Bone marrow evaluation revealed a hypercellular marrow with megakaryocytic and myeloid dysplasia with increased marrow fibrosis, less than 5% myeloid blasts, and complex cytogenetics. NGS analysis of bone marrow detected a TP53 mutation c.524G>A with a variant allele frequency (VAF) of 72.4%. A custom ddPCR assay detected this mutation in blood before, and then consistently increasing after, AlloHCT (Figure 1). The patient received 3 cycles of decitabine with progression followed by 1 cycle of azacytidine. He died shortly thereafter from severe pulmonary hypertension at 3 years posttransplant.
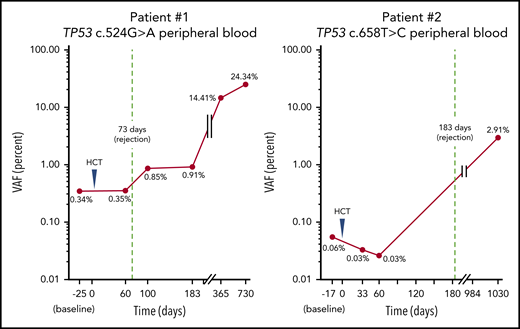
TP53 mutations are detectable in blood before transplantation and increase until therapy-related myeloid malignancy diagnosis. Mutations detected in bone marrow at time of myeloid malignancy diagnosis using NGS could be tracked in blood more than 2 years earlier using ddPCR.
TP53 mutations are detectable in blood before transplantation and increase until therapy-related myeloid malignancy diagnosis. Mutations detected in bone marrow at time of myeloid malignancy diagnosis using NGS could be tracked in blood more than 2 years earlier using ddPCR.
A second patient had HbSS complicated by frequent VOC and chronic pain. He underwent HLA-matched sibling PBSCT at 37 years of age and underwent conditioning with alemtuzumab and 300 cGy total-body irradiation. He initially engrafted but then rejected his graft at 6 months posttransplant. Bone marrow examination at that time demonstrated hypoplasia, a complex karyotype, but no blasts or dysplasia. Two and half years posttransplant, he presented with worsening anemia and thrombocytopenia. A repeat bone marrow evaluation now showed hyperplasia without evidence of increased blasts. Peripheral blood smear revealed evidence of dyserythropoiesis. NGS analysis of bone marrow demonstrated a TP53 mutation c.658T>C at a VAF of 4.5%. A custom ddPCR assay also detected this mutation in blood before initial transplantation (Figure 1). The patient underwent salvage myeloablative haploidentical PBSCT 3 years after his first transplant, but died 47 days later of intracranial hemorrhage.
A third patient also developed therapy-related myeloid neoplasm 5 years after AlloHCT. She had been conditioned with alemtuzumab and 400 cGy total-body irradiation. Bone marrow evaluation at an outside facility revealed a hypercellular diffusely fibrotic marrow with dysplastic megakaryocytic hyperplasia and 10% to 15% blasts. Cytogenetic results revealed a 7q deletion. NGS was not performed at this time, and no samples were available for laboratory evaluation.
Consistent with individual case reports from other institutions,8,16 development of myeloid malignancy was only seen in those rejecting their grafts. None of the 57 patients who engrafted developed MDS/AML compared with these 3 cases out of 19 patients who rejected their grafts. Further, no TP53 mutations were detected by NGS in pretransplant bone marrow samples from 6 patients with HbSS undergoing AlloHCT: 4 who rejected their grafts are between 4.4 and 7.7 years and 2 engrafted patients with mixed myeloid chimerism are between 6.4 and 6.8 years post-AlloHCT. None has developed a myeloid malignancy. Further studies should evaluate the effect of graft rejection and mixed chimerism on the incidence of myeloid malignancies post-AlloHCT.
In 2 patients with SCD, we report clonal expansion of TP53 mutations present at the time of myeloid malignancy diagnosis from earlier points, including before initial transplantation 2 to 3 years earlier. Wong and colleagues identified 4 patients with therapy-related MDS/AML, and TP53 mutations had the same mutations existent at low frequencies (0.003%-0.7%) 3 to 6 years before.17 Further, they showed in mice that received bone marrow mixtures of wild-type and TP53-mutated hematopoietic stem/progenitor cells that the TP53 mutated clones specially expanded after chemotherapy exposure. Because of factors including erythropoietic stress and systemic inflammation, our patients may have been predisposed to developing clonal hematopoiesis, which is not typically seen until older age.18,19 As these clones may be more resistant to radiation and/or chemotherapy, they may preferentially expand after a failed transplant, leading to the myeloid malignancy seen in our patients who rejected their grafts.
Although all 3 patients were treated with hydroxyurea before transplant, the exact duration of therapy is unknown. And because we do not have prehydroxyurea blood and bone marrow samples, we cannot comment on the association between hydroxyurea treatment and development of TP53-mutated clones. Hydroxyurea was first approved by the US Food and Drug Administration for SCD more than 2 decades ago, and a clear increased risk for MDS/AML has not been demonstrated. Although case reports describe patients who developed MDS/AML after short- and long-term hydroxyurea usage, larger studies have not revealed a significant increase in the risk for hematologic malignancies in patients with hydroxyurea exposure.2,20 Further, data regarding whether hydroxyurea is associated with genotoxicity are conflicting.21,22
In summary, we report for the first time in patients with SCD after unsuccessful alloHCT the progression of baseline high-risk TP53 clonal abnormalities into myeloid malignancy. This rare complication occurred only in patients who did not engraft, and multiple trials are ongoing with the goal of decreasing the graft rejection rate in patients with SCD. Detectable mutations before reduced-intensity AlloHCT in patients with AML in remission is associated with posttransplant relapse of myeloid malignancy.23 The predictive value and clinical utility of screening for myeloid malignancy-associated mutations such as TP53 in patients with HbSS before AlloHCT is, however, currently unknown. Long-term follow-up of all patients with SCD post-AlloHCT is critical, and larger studies are indicated.
FASTQ files are available in the National Center for Biotechnology Information Small Reads Archive (accession no. PRJNA606770).
Acknowledgment
This work was funded by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health.
Authorship
Contribution: J.Y.G., X.X., and L.D. performed the experiments and assisted with data analyses and in manuscript preparation; I.M. and M.M.H. assisted with data collection and in manuscript preparation; Y.L. assisted with laboratory experiments and in manuscript preparation; and C.S.H. and C.D.F. designed the study, analyzed the data, and wrote the manuscript
Conflict-of-interest disclosure: C.S.H. receives laboratory research funding from Merck and Sellas. The remaining authors declare no competing financial interests.
Correspondence: Courtney D. Fitzhugh, Laboratory of Early Sickle Mortality Prevention, Cellular and Molecular Therapeutics Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Dr, MSC 1589, Building 10, Room 6N240A, Bethesda, MD 20892; e-mail: fitzhughc@nhlbi.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal