Key Points
In the context of gene therapy, the estimated number of active, repopulating HSPCs was correlated with the number of HSPCs per kilogram infused.
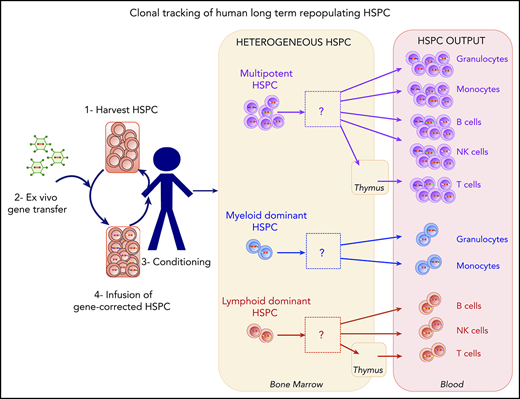
An analysis of human HSPC clonal lineage outputs highlighted the presence of myeloid-dominant, lymphoid-dominant, and balanced cell subsets.

Abstract
In gene therapy with human hematopoietic stem and progenitor cells (HSPCs), each gene-corrected cell and its progeny are marked in a unique way by the integrating vector. This feature enables lineages to be tracked by sampling blood cells and using DNA sequencing to identify the vector integration sites. Here, we studied 5 cell lineages (granulocytes, monocytes, T cells, B cells, and natural killer cells) in patients having undergone HSPC gene therapy for Wiskott-Aldrich syndrome or β hemoglobinopathies. We found that the estimated minimum number of active, repopulating HSPCs (which ranged from 2000 to 50 000) was correlated with the number of HSPCs per kilogram infused. We sought to quantify the lineage output and dynamics of gene-modified clones; this is usually challenging because of sparse sampling of the various cell types during the analytical procedure, contamination during cell isolation, and different levels of vector marking in the various lineages. We therefore measured the residual contamination and corrected our statistical models accordingly to provide a rigorous analysis of the HSPC lineage output. A cluster analysis of the HSPC lineage output highlighted the existence of several stable, distinct differentiation programs, including myeloid-dominant, lymphoid-dominant, and balanced cell subsets. Our study evidenced the heterogeneous nature of the cell lineage output from HSPCs and provided methods for analyzing these complex data.
Introduction
Hematopoietic stem cells (HSCs) are defined by their ability to self-renew while producing daughter cells capable of differentiation, and thus enabling the sustained production of all blood cell lineages. Literature data from in vitro differentiation and transplantation assays in murine models have suggested that HSCs differentiate into multipotent progenitors, which in turn give rise to early committed progenitors that progressively lose their self-renewal ability. The early committed progenitors segregate into common myeloid progenitors and common lymphoid progenitors.1,2 However, this classical model has been challenged by the identification of other self-renewing progenitors, including lymphomyeloid-restricted progenitors (ie, cells having lost their megakaryocyte and erythroid potential) and myeloid-restricted progenitors (ie, cells having retained their long-term myeloid and megakaryocyte potential).3-7 Cells may thus lose their multipotency while retaining the ability to self-renew and produce a restricted number of lineages.8 The classical model has been further challenged by the documented heterogeneity of murine HSC self-renewal and reconstitution,9 and the identification of stem cells that can give rise to cell populations with different myeloid:lymphoid ratios.5,10,11 Most recently, the combination of genetic barcoding and labeling methods with murine transplantation studies has increased the accuracy of clonal tracking and confirmed the existence of discrete HSC subsets12-16 and multilineage/oligolineage HSC clones.17
A clonal tracking study of lentiviral integration sites (ISs) in macaques documented the existence of 3 groups of HSCs with different myeloid and lymphoid potentials.18 In the same nonhuman primate model, Dunbar's group recently used a quantitative barcoding approach to observe relatively stable, multipotent, long-term, clonal HSC outputs, together with clones whose output was biased toward myeloid or lymphoid lineages.19,20 Taken as a whole, the results of animal studies suggest that long-lived clones can be subdivided into several functional groups.
In humans, decades of therapeutic stem cell transplantation have shown that long-term repopulating HSCs are part of the CD34+ subset or (according to some studies) the CD133+ cell subset21 that comprise a mixture of hematopoietic stem and progenitor cells (HSPCs). Xenotransplantation in immunodeficient nonobese diabetic-severe combined immunodeficiency gammaC−/− mice can be used as a surrogate to distinguish between committed progenitors on one hand and HSCs capable of long-term engraftment on the other.22 Barcoding analyses of human CD34+ HSPCs engrafted in nonobese diabetic-severe combined immunodeficiency gammaC−/− mice also suggest that the HSPC potential is heterogeneous in humans.23,24 However, the long-term repopulation capacity is limited by the animal’s life span, and the interpretation of these data in mice is complicated by a skewing of human cell differentiation toward lymphoid lineages.
Human gene therapy based on the ex vivo transduction of CD34+ cells with an integrating vector provides an opportunity to directly track stem cell activity in humans.25 Integration of the therapeutic vector marks the genome at unique positions in each cell, and this mark is transmitted to the cell’s progeny. Thus, tracking ISs in fractionated blood cell lineages enables the clonal tracking of stem cell progeny. Initial reports on gene therapy trials for Wiskott-Aldrich syndrome (WAS) and metachromatic leukodystrophy26,27 showed that lymphoid lineages, myeloid lineages, and bone marrow (BM) CD34+ cells shared ISs. Recently, Biasco et al performed a more detailed analysis of clonal dynamics in 4 patients from a WAS gene therapy trial by taking advantage of marking with integrated vectors. Their data suggested that reconstitution had occurred in 2 waves, with a 12-month interval between cell transplantation and the establishment of steady-state hematopoiesis.28,29 Hence, the myeloid and lymphoid lineages appeared to have segregated relatively late in development. This pioneering study provided the most in-depth look to date of human hematopoiesis as revealed by IS tracking in gene therapy patients.
To investigate human HSPC function in more detail, we applied IS mapping to track HSPC dynamics in 6 patients from 2 gene therapy trials in which lentiviral vectors had been used to introduce copies of healthy genes. We extended Biasco et al’s study by analyzing more than 1 disease, quantifying residual cell contamination and differences in sampling between cell lineages, and generating a statistical model of the quantitative clonal lineage output. It may be difficult to draw firm conclusions about the biology of human hematopoietic cells when the corrected cells may or may not have a selective advantage over their noncorrected counterparts; hence, studies of patients with different disorders are preferable. Here, we analyzed 4 patients treated for WAS,30 1 patient treated for sickle cell disease (βS/βS),31 and 1 patient treated for β thalassemia (β0/βE).32 Peripheral blood samples were fractionated into 5 blood cell lineages, and HSPC dynamics and lineage outputs were tracked by using vector ISs as markers.
Methods
Patients
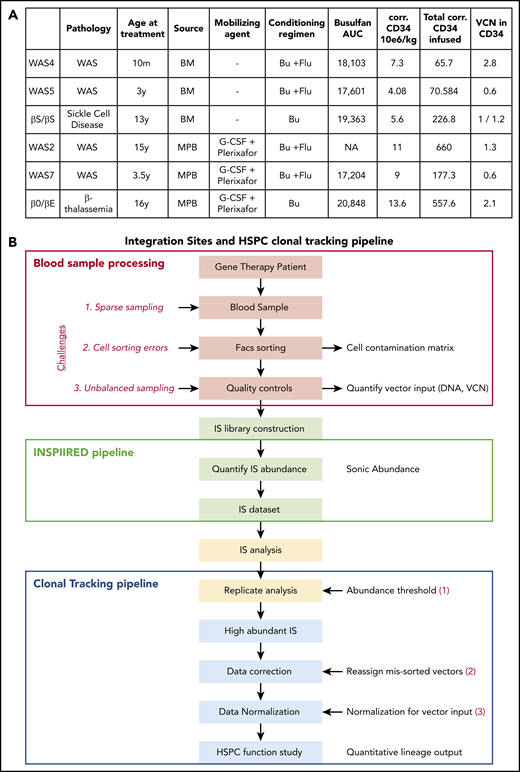
Four patients with WAS were included in the present study and have been described previously as part of a gene therapy trial.30 The patient designations used here are the same as in the earlier publication. The 2 patients with β hemoglobinopathy (1 with βS/βS sickle cell disease31 and 1 with β0/βE β thalassemia32 ) were participating in the HGB205 clinical trial and were chosen because of their long follow-up period. Autologous HSPCs were derived from BM or from mobilized peripheral blood (MPB; Figure 1A) collected by apheresis after the administration of granulocyte colony-stimulating factor and the CXCR4 antagonist plerixafor.33 All patients received a myeloablative conditioning regimen that promoted the engraftment of gene-corrected HSPCs. Samples were obtained through gene therapy protocols set up in the Biotherapy Clinical Investigation Center at Necker Children’s Hospital (Paris, France). All gene therapy and follow-up protocols were approved by the local institutional review board (CPP Ile-de-France II, Paris, France; reference for WAS: 2014-04-02-MS1; reference for β thalassemia and sickle cell disease: 2013/35). The number of corrected HSPCs infused was determined by multiplying the number of CD34+ cells infused in the patient by the vector copy number (VCN) measured in CD34+ cells (for VCN < 1).
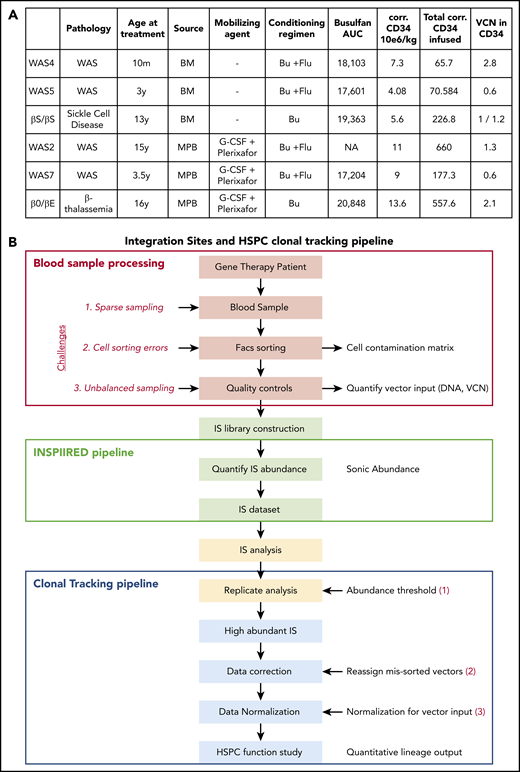
HSPC lineage tracking in human gene therapy trials. (A) Characteristics of the gene therapy patients studied here. AUC, area under the curve; Bu, busulfan; corr.: corrected; Flu, fludarabine; y: year; m: month. (B) Workflow for the IS clonal tracking analysis, from patient blood sample processing, IS library construction, and IS data preparation to functional studies of the HSPCs.
HSPC lineage tracking in human gene therapy trials. (A) Characteristics of the gene therapy patients studied here. AUC, area under the curve; Bu, busulfan; corr.: corrected; Flu, fludarabine; y: year; m: month. (B) Workflow for the IS clonal tracking analysis, from patient blood sample processing, IS library construction, and IS data preparation to functional studies of the HSPCs.
Identification and quantification of ISs
Integration sites were amplified for sequencing and analyzed using the INSPIIRED pipeline, as described previously.34 The sites were isolated from each patient’s genomic DNA, using nested ligation-mediated polymerase chain reaction (PCR) after unbiased fragmentation (Covaris System). The samples were tracked by dual indexing, cycling through variations of 96 linkers, and error-correcting the barcodes. Sample and library concentrations were quantified using a KAPA SYBR FAST Universal qPCR Kit, and libraries were pooled on the basis of the sample measurements. Libraries were diluted or concentrated to 1 to 4 nM before the Miseq loading protocol. AMPure XP beads were used to purify and concentrate the DNA. Sequenced reads were aligned with the hg38 reference genome (>95% identity), using BLAT (BLAST-like alignment tool v35). Integration sites marked single clones whose clonal abundances were determined with the SonicAbundance method; the length of the DNA fragments flanking ISs is used to document independent isolations of integration events, and thus provides an estimate of the number of cells associated with each unique IS.34-36 Multiple replicates (4-20) of each sample were analyzed independently to reduce founder effects during PCR, and the stochastic sampling. PCR contamination during library preparation can be a source of error. To suppress PCR crossover, each sheared DNA sample was ligated to a unique linker. A dual barcoding strategy was then used to filter out PCR crossovers.34 To monitor possible PCR contamination, a total of 41 control samples of human DNA lacking ISs were analyzed in parallel with the patient samples. After sequence acquisition and analysis, 40 control samples did not show any IS, and 1 sample showed 2 ISs. We concluded that PCR contamination was not a significant confounder in our analysis.
Full details of methods for other procedures are provided in the supplemental Methods, available on the Blood Web site.
Results
Clonal tracking in patients after gene therapy with integrating vectors
To determine the activity of vector-modified progenitor cells, we analyzed 4 patients treated for WAS30 and 2 patients treated for β hemoglobinopathies31,32 (Figure 1A; the patients’ reference numbers are the same as in the primary publications). All had successfully undergone gene therapy with integrating lentiviral vectors. Autologous HSPCs were derived from either BM (WAS4, WAS5, and the βS/βS patient) or MPB (WAS2, WAS7, and the β0/βE patient; Figure 1A).
For each patient, 5 cell types were sorted from peripheral blood samples: granulocytes, monocytes, T cells, B cells, and natural killer (NK) cells (referred to as G, M, T, B, and K, respectively; supplemental Figure 1). DNA from these samples was analyzed to determine the IS distribution and their clonal abundance, using the INSPIIRED pipeline and the sonicAbundance method.34-36 Each patient provided samples for at least 2 points, with the first around 1 year after gene correction and the last at least 2 years after gene correction (Figure 2). The patients showed gene marking in all the blood cell lineages analyzed, as evidenced by the VCN (supplemental Figure 2A).
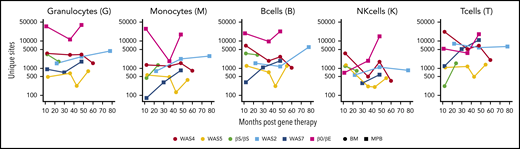
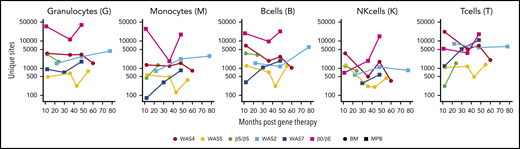
Numbers of unique ISs in gene-corrected patients. The number of unique ISs for granulocytes, monocytes, B cells, NK cells, and T cells at various points (in months) after gene therapy. Patients received HSPCs sourced from BM (circles) or MPB (squares).
Numbers of unique ISs in gene-corrected patients. The number of unique ISs for granulocytes, monocytes, B cells, NK cells, and T cells at various points (in months) after gene therapy. Patients received HSPCs sourced from BM (circles) or MPB (squares).
At the latest time, the number of unique ISs detected per patient ranged from 2941 (in the WAS5 patient) to 98 185 (in the patient with β0/βE; supplemental Table 1). The number of ISs per cell type varied (Figure 2) with the level of gene marking (ie, the VCN) and the amount of DNA used to build the library (supplemental Figure 2).
We observed high clonal diversity for ISs within the sampled cell types, as measured by the Shannon diversity index37 (supplemental Figure 3). The diversity values increase with an increase in the numbers of ISs or an increase in the evenness of distribution; hence, a high degree of diversity reflects successful polyclonal gene correction.
To gain an overview of the IS distributions, we first mapped the sites relative to genomic features (supplemental Figure 4A) and epigenetic marks (supplemental Figure 4B). In all 5 cell lineages, the vector integrated preferentially within transcription units and gene-rich regions and near marks associated with active transcription, as expected for lentiviral vectors.38-40 There was no obvious selection during hematopoietic reconstitution (ie, there were no increases in integration frequency near particular annotated chromosomal features).
To check for clonal skewing caused by insertional mutagenesis, we next looked for increased frequencies of clones harboring ISs near specific genes. To this end, we compared pooled data from the earliest point with pooled data from the last point (supplemental Figure 4C). No obvious selective clonal outgrowth was detected. Thus, if clonal skewing had taken place, it had not strongly affected proliferation between the first and last points. We did not observe an increase over time in the frequency of integration near cancer-associated genes (supplemental Table 2).
Estimation of the minimum number of active HSPCs capable of long-term engraftment
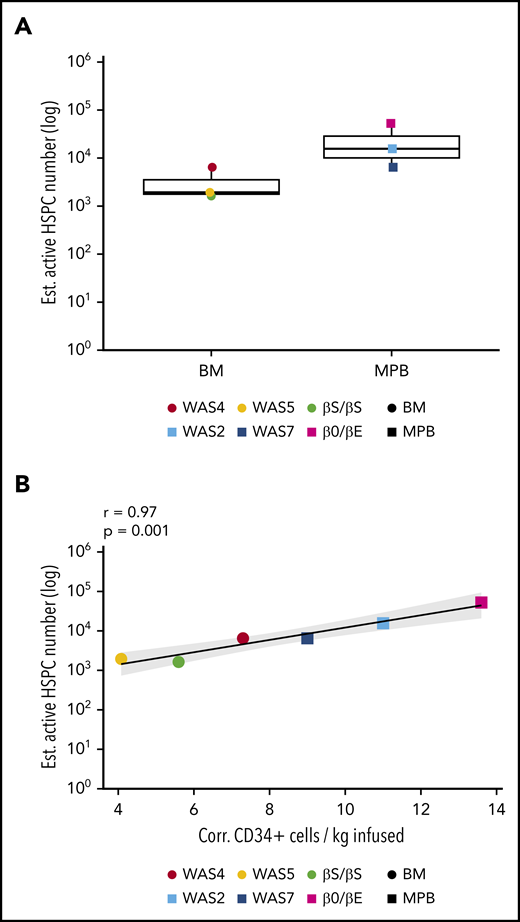
To estimate the minimum true population size of gene-corrected and active HSPCs, we used the Chao1 estimator.41 The latter provides an estimate of the minimum total number of unique ISs, and thus the minimum total number of gene-corrected cells (see the supplemental Methods for more details). We concentrated on the latest point to estimate the populations of long-term repopulating cells and granulocytes; because of their short lifespans (1-5 days), these populations best reflect HSPC dynamics.42 The estimated minimum population of active, granulocyte-generating HSPCs ranged from 1943 to 52 444 (Figure 3A).
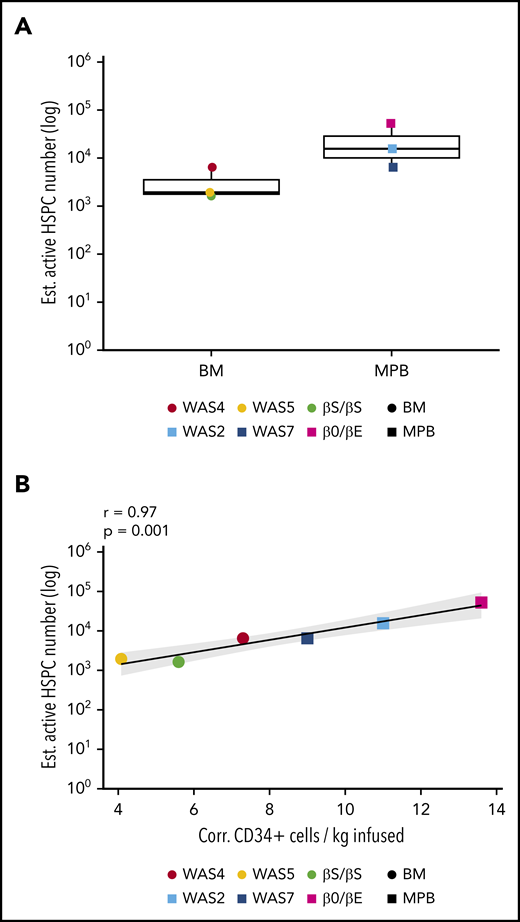
Estimation of the number of active HSPCs. (A) The estimated number of active, long-term-repopulating HSPCs, corresponding to the estimated granulocyte population size at last follow-up with a normalized Chao1 estimate (the Chao1 estimate divided by VCN for VCN >1; supplemental Methods). (B) The correlation between the number of active, long-term-repopulating HSPCs (Chao1 estimate of the size of the granulocyte population) and the number of corrected CD34+ cells infused per kilogram for each patient.
Estimation of the number of active HSPCs. (A) The estimated number of active, long-term-repopulating HSPCs, corresponding to the estimated granulocyte population size at last follow-up with a normalized Chao1 estimate (the Chao1 estimate divided by VCN for VCN >1; supplemental Methods). (B) The correlation between the number of active, long-term-repopulating HSPCs (Chao1 estimate of the size of the granulocyte population) and the number of corrected CD34+ cells infused per kilogram for each patient.
The estimated minimum frequency of active, long-term engrafting clones producing granulocytes ranged from 0.0007% to 0.01% of the initial corrected CD34+ cell population; this corresponded to 7 to 98 repopulating HSPCs per 106 infused CD34+ cells (supplemental Figure 5). We observed a positive correlation between the estimated number of active HSPCs in each patient and the total number of corrected CD34+ cells infused (data not shown), but the highest correlation was detected with the number of corrected CD34+ cells per kilogram infused (Figure 3B).
Analysis of highly active gene-modified clones
To model HSPC activity using IS data, one would ideally reconstruct each clone’s cell lineage output from the sequencing data specified by the IS positions. However, the data generation process involves several steps that add uncertainty to the estimated population compositions. We therefore used statistical techniques to model the total populations (Figure 1B; supplemental Methods).
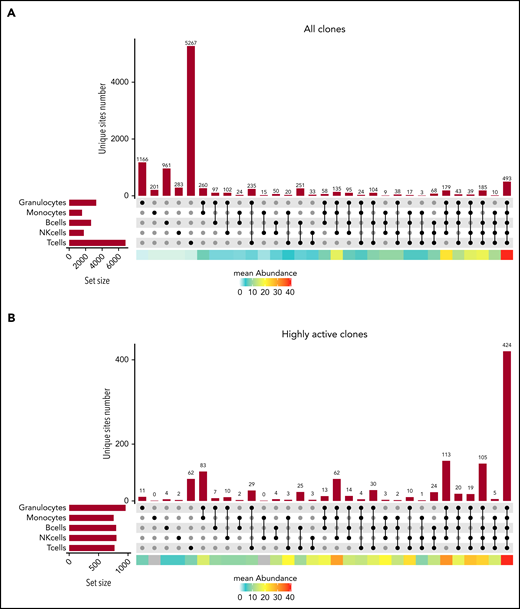
Quantifying the ISs present in combinations of cell lineages (Figure 4A; supplemental Figure 6A) revealed clones from all 5 lineages (493 for WAS4 at m48). However, a large proportion of the clones was present at a low abundance in a single lineage (eg, the 5267 clones detected in T cells only for WAS4 m48). To assess the efficiency of our method for IS identification, we compared the results from library technical replicates and cell sorting biological replicates.19 We found that many IS were present in a single replicate only, which highlights the challenge posed by sparse sampling for this type of analysis (supplemental Figures 7-9). Hence, in subsequent analyses, we focused on highly active clones detected consistently at high levels in the replicates. An assessment of the number of cells sampled showed that clones comprising at least 6 cells were shared at 97% between replicates.
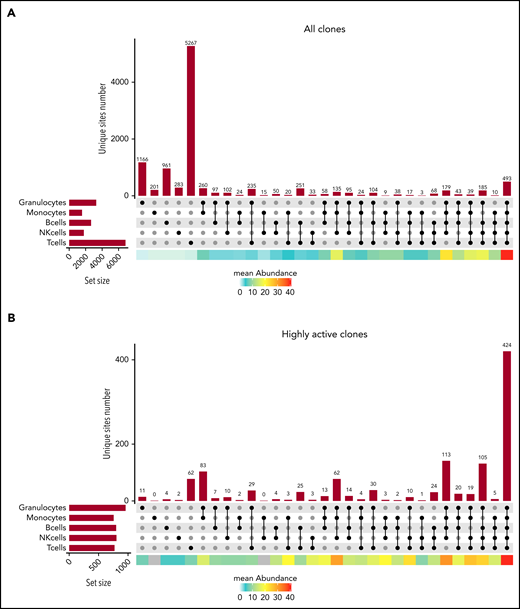
Clonal detection of ISs in the various cell lineages. (A) An UpSet plot showing the number of ISs detected in each single lineage and any combination of 2, 3, 4, or 5 cell lineages for patient WAS4 at m48 of follow-up, considering the IS dataset as a whole. The mean abundance in each category is shown by the heat map at the bottom of the graph. (B) An UpSet plot showing the number of ISs detected in each single lineage and any combination of 2, 3, 4, or 5 cell lineages for patient WAS4 at m48 of follow-up, considering only the highly abundant ISs. The mean abundance in each category is shown by the heat map at the bottom of the graph.
Clonal detection of ISs in the various cell lineages. (A) An UpSet plot showing the number of ISs detected in each single lineage and any combination of 2, 3, 4, or 5 cell lineages for patient WAS4 at m48 of follow-up, considering the IS dataset as a whole. The mean abundance in each category is shown by the heat map at the bottom of the graph. (B) An UpSet plot showing the number of ISs detected in each single lineage and any combination of 2, 3, 4, or 5 cell lineages for patient WAS4 at m48 of follow-up, considering only the highly abundant ISs. The mean abundance in each category is shown by the heat map at the bottom of the graph.
Analysis of well-sampled clones revealed a diversity of compositions: although most of the clones (n = 424 for WAS4 m48) were detected in all 5 lineages, a significant number of clones were detected in 2 or 1 lineages (n = 83 in G and M; n = 62 in T for WAS4 m48; Figure 4B; supplemental Figure 6A). Importantly, we found that the detection of ISs in 4 or 5 cell lineages is not sufficient to define a multipotent HSPC lineage output, as there were sometimes large differences in abundance between lineages (supplemental Figure 6B). Thus, a quantitative investigation of cell lineages is required to discriminate between a multipotent lineage output and a biased lineage output.
A quantitative investigation should also take account of errors introduced during cell sorting and interlineage differences in sampling (as a result of varying levels of gene marking in each lineage and varying amounts of cellular DNA available for IS analysis; supplemental Figure 2). In our pipeline, we corrected for both sources of error (supplemental Methods; supplemental Table 4); this enabled us to model the cell lineage proportions produced by the most active progenitors within a rigorous statistical framework.
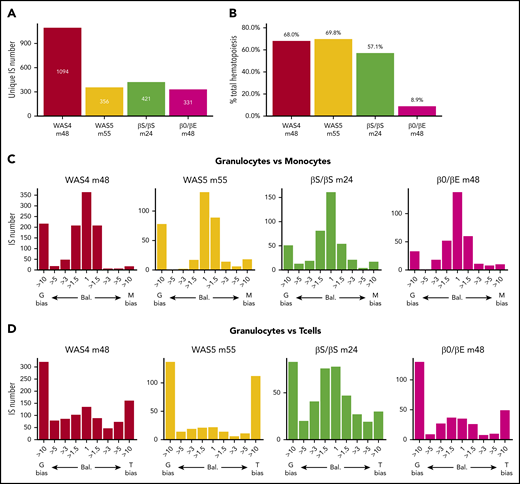
Lineage bias among active HSPCs
In the following analysis, we focused on the corrected data for 4 patient/time combinations with the most active clones (WAS4, WAS5, βS/βS, and β0/βE at m48, m55, m24, and m48, respectively; Figure 5A). This corresponded to 331 to 1094 highly active clones (corresponding to 8% to 12% of all the ISs for the patients WAS4, WAS5, and βS/βS and 0.3% for the β0/βE patient). The clones contributed between 9% and 69% of the total hematopoietic output (Figure 5B). We first evaluated lineage bias by analyzing Pearson’s correlation coefficient for corrected clonal abundances in pairs of cell types (supplemental Figure 10A). We observed the strongest correlations for granulocytes vs monocytes (0.77 to 0.97), intermediate correlations for granulocytes vs B cells (0.31 to 0.62) and for T cells vs B cells (−0.01 to 0.67), and the lowest correlations for T cells vs the other cell lineages (−0.011 to 0.47).
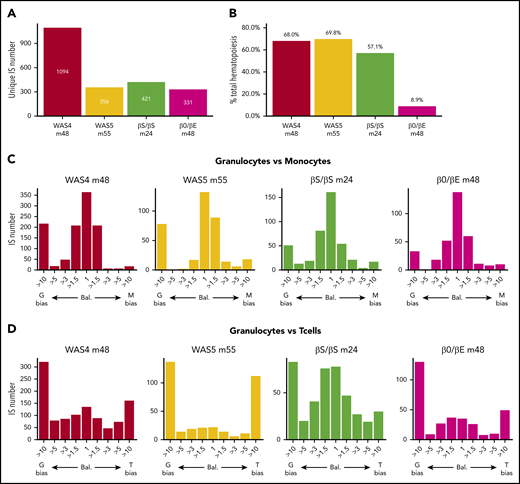
Clonal contributions to various hematopoietic cell types. (A) The number of highly active HSPC clones for each patient at last follow-up (unique ISs with at least 6 cells in 1 of the 5 cell types). (B) Contribution of highly active clones to total hematopoiesis output. The proportion of the total cell count (from all cell types) in the whole IS dataset accounted for by highly active clones is shown. (C-D) Lineage bias analysis for different combinations of cell lineage abundance and in each patient, represented as a bias histogram. For each IS, the ratio of abundance is calculated for the 2 cell types and then classified as balanced (Bal.) through a >10-fold difference. (C) Lineage bias for granulocytes vs monocytes (G vs M). (D) Lineage bias for granulocytes vs T cells (G vs T).
Clonal contributions to various hematopoietic cell types. (A) The number of highly active HSPC clones for each patient at last follow-up (unique ISs with at least 6 cells in 1 of the 5 cell types). (B) Contribution of highly active clones to total hematopoiesis output. The proportion of the total cell count (from all cell types) in the whole IS dataset accounted for by highly active clones is shown. (C-D) Lineage bias analysis for different combinations of cell lineage abundance and in each patient, represented as a bias histogram. For each IS, the ratio of abundance is calculated for the 2 cell types and then classified as balanced (Bal.) through a >10-fold difference. (C) Lineage bias for granulocytes vs monocytes (G vs M). (D) Lineage bias for granulocytes vs T cells (G vs T).
To investigate these differences further, we evaluated abundance ratios and potential bias toward certain cell types (defined as at least a 10-fold difference between a pair of cell types19 ). For each pair of cell types, ISs can variously be assigned to balanced clones (ie, bias = 1) or clones with more than a 10-fold bias toward one lineage or another (Figure 5C-D; supplemental Figure 10B). We mostly detected balanced contributions for granulocytes vs monocytes. However, our analysis of granulocytes vs T cells revealed that the majority of clones were biased toward one cell type, and few clones had a balanced contribution. The long half-life of lymphoid cells (relative to granulocytes) might complicate the interpretation of this result by favoring the accumulation of a lymphoid-biased population. However, an analysis of pooled data from the 4 available points for WAS4 gave similar results (supplemental Figure 11), and thus emphasized the heterogeneity of the HSPC lineage output.
Identification of distinct HSPC subsets
We next analyzed the human HSPC lineage output in the 5 cell lineages for each highly active clone and estimated the lineage potential. In an initial analysis, each IS was quantitatively mapped to 1 of the 31 reference cell type compositions containing all combinations of 1, 2, 3, 4, or 5 cell types (supplemental Figure 12). A Kullback-Leibler divergence analysis was used to define the closest reference composition (supplemental Methods), and it revealed a broad variety of cell compositions, ranging from unipotent clones to clones contributing to all 5 lineages.
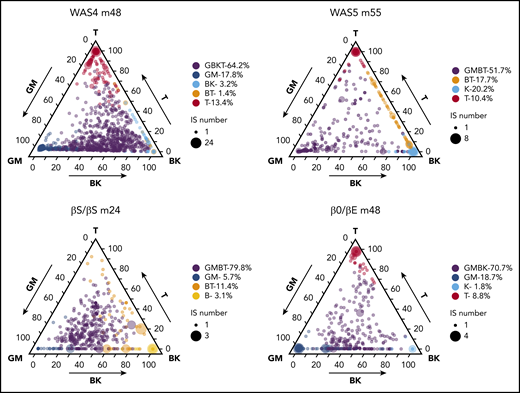
We next looked for possible HSPC subtypes via unsupervised K-means clustering of the data (supplemental Methods). Each patient displayed 4 to 6 predominant clusters, each of which was characterized by a specific lineage composition (potentially corresponding to a class of HSPC). Ternary plots showed the various IS clones’ respective lineage outputs (Figure 6; supplemental Figure 13). The lineage composition of each class is described by the group’s centroid (supplemental Figure 14).
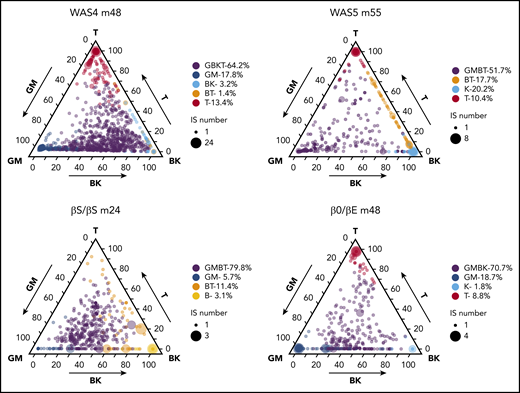
HSPC subsets and HSPC heterogeneity. Ternary plots for each patient, showing the K-means cluster analysis of highly active ISs and highlighting the presence of 4 to 6 predominant cell type compositions (HSPC subsets). The lineage output inferred for each IS clone is projected onto a triangle that combines the 5 cell lineages and 3 axis: GM/T/BK (with the proportion of granulocytes and monocytes on the left axis, the proportion of T cells on the right axis, and the proportion of B+NK cells on the bottom axis). The corrected relative abundance of each clone at a specific position is indicated by the dot density. The total number of IS clones at a specific position is indicated by the dot size. The clusters are named according to their centroid (shown in supplemental Figure 14), using the closest reference composition (according to the minimum Kullback-Leibler distance to the 31 reference compositions; supplemental Methods). The proportions of IS in each cluster related to the total number of IS clones are indicated.
HSPC subsets and HSPC heterogeneity. Ternary plots for each patient, showing the K-means cluster analysis of highly active ISs and highlighting the presence of 4 to 6 predominant cell type compositions (HSPC subsets). The lineage output inferred for each IS clone is projected onto a triangle that combines the 5 cell lineages and 3 axis: GM/T/BK (with the proportion of granulocytes and monocytes on the left axis, the proportion of T cells on the right axis, and the proportion of B+NK cells on the bottom axis). The corrected relative abundance of each clone at a specific position is indicated by the dot density. The total number of IS clones at a specific position is indicated by the dot size. The clusters are named according to their centroid (shown in supplemental Figure 14), using the closest reference composition (according to the minimum Kullback-Leibler distance to the 31 reference compositions; supplemental Methods). The proportions of IS in each cluster related to the total number of IS clones are indicated.
In all 4 patients, at least half of the active clones (52%-80%) clustered within a group whose centroid composition was close to GMBT, GBKT, or GMBK (in purple, Figure 6), as would be expected for multipotent HSPCs. The analysis also revealed another significant lymphoid-dominant group of clones in 3 patients (ie, leading almost exclusively to the production of T cells), which appeared at the ternary plot’s T apex (in red in Figure 6). This group accounted for 9% to 13% of the IS clones and was detected in all patients other than the βS/βS individual (in whom a BT group was detected instead). The next most frequent group (accounting for 6% to 19% of IS clones in 3 patients) corresponded to myeloid-dominant clones, which appeared at the ternary plot’s GM apex (in blue in Figure 6). In the remaining groups of clones, we noted an NK group (in turquoise) in 2 patients that accounted respectively for 2% and 20% of the clones. Thus, the results of the K-means cluster analysis suggested the coexistence of myeloid-dominant and lymphoid-dominant (mostly T-dominant) HSPC subsets and more balanced, multipotent HSPC subsets in each patient. Further longitudinal sampling will be required to determine whether the absence of a myeloid-dominant subset (in WAS5) or a T-dominant subset (in βS/βS) corresponds to interpatient heterogeneity or was a result of the smaller number of clones analyzed (relative to WAS4).
To further characterize these distinct HSPC clones, we quantified and compared their ability to produce granulocytes and T cells (supplemental Figure 15). The granulocyte abundance was similar in the myeloid-dominant group and multipotent groups (WAS4), or was slightly lower in the myeloid-dominant group than in the multipotent groups (βS/βS and β0/βE; supplemental Figure 15A). The T-cell abundance was similar in the T-cell-dominant group (WAS5) or was slightly higher in the T-cell-dominant group than in the multipotent groups (supplemental Figure 15B). These results suggest that multipotent and lineage bias clones produced their progeny with a very similar level of efficiency.
Last, we developed a modeling approach and used it to assess our clonal tracking pipeline’s limit of detection (supplemental Figure 16A; supplemental Methods). We simulated the whole blood population containing gene-marked cells (supplemental Figure 16B) as either a homogeneous multipotent population or a heterogeneous population containing 3 HSPC subsets (60% multipotent HSPC, 20% myeloid-dominant HSPC, and 20% T-cell dominant HSPC), to which the errors having occurred during sample processing were applied. The results of the simulation indicated that consideration of all detected clones (supplemental Figure 16C) can give rise to sparse sampling, and thus the false detection of lineage-biased clones, as shown by our lineage bias analysis of granulocytes vs T cells and our cluster analysis. In this sampled population, it was not possible to distinguish between the 2 HSPC configurations. In contrast, highly abundant clones (supplemental Figure 16D) closely resemble the whole-blood population (supplemental Figure 16B) in terms of both lineage bias and clustering. Concentrating on highly abundant clones enables one to discriminate between homogeneous and heterogeneous HSPC populations.
By combining an HSPC lineage output analysis with a simulation approach, we were able to highlight the heterogeneity of human HSPCs (ie, the coexistence of multipotent, myeloid-dominant, and lymphoid-dominant HSPC subsets).
Changes over time in HSPC subsets
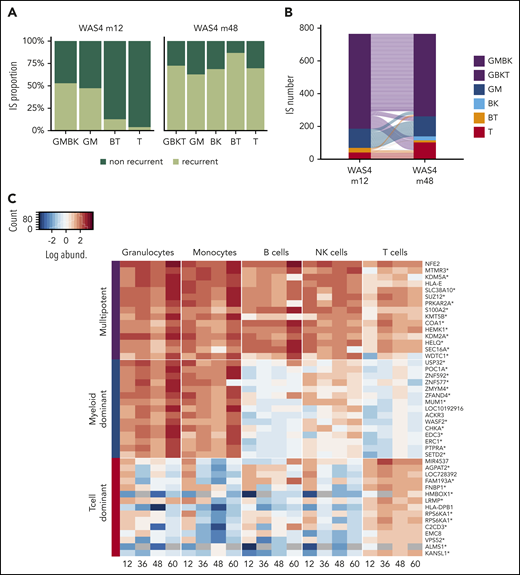
To investigate changes over time in HSPC clones, we focused on patient WAS4; 4 points were available, and m12 and m48 were sufficiently well sampled (with good correlation between replicates; supplemental Figure 9) to allow reliable cluster analysis. At m12, 2652 highly abundant clones accounted for 56% of the total cell count (supplemental Figure 17A-B). A cluster analysis revealed the presence of multipotent, T-cell-dominant, and myeloid-dominant clones (supplemental Figure 17C). An analysis of clones present at m12 and m48 showed that the proportion of multipotent clones did not change markedly over time (52% at m12 and 72% at m48; Figure 7A). The same was true of the myeloid-dominant clones. In contrast, a large proportion of the T-lymphoid-dominant clones present at m12 (96%, corresponding to 1035 IS) were not present at m48 (Figure 7A), and might therefore correspond to progenitors lacking the ability to self-renew.
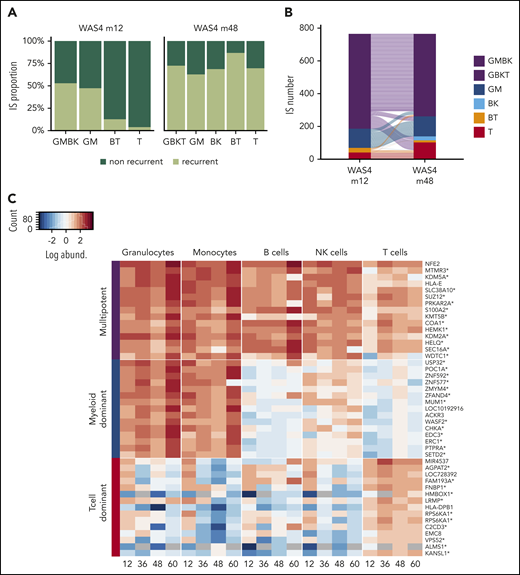
Dynamic of HSPC subsets. (A) Proportion of ISs persisting over time in each IS subset. (B) An alluvial plot showing the changes in the different IS subsets comprising the 768 clones that were present at both m12 and m48. (C) A heat map showing the TOP15 IS clones from multipotent, myeloid-dominant, and T-cell-dominant subsets at m48 for the 4 available points (m12, m36, m48, and m60). Each row represents a unique IS clone, with its closest gene labeled on the right. Integration within the transcription unit is indicated by a star. IS abundance is represented by color-code scale.
Dynamic of HSPC subsets. (A) Proportion of ISs persisting over time in each IS subset. (B) An alluvial plot showing the changes in the different IS subsets comprising the 768 clones that were present at both m12 and m48. (C) A heat map showing the TOP15 IS clones from multipotent, myeloid-dominant, and T-cell-dominant subsets at m48 for the 4 available points (m12, m36, m48, and m60). Each row represents a unique IS clone, with its closest gene labeled on the right. Integration within the transcription unit is indicated by a star. IS abundance is represented by color-code scale.
We next sought to determine whether the lineage potential of the various clonal subsets were stable over time by concentrating on the fraction of clones shared between m12 and m48. We found that the majority of multipotent clones at m12 (81% GMBK) were multipotent at m48 (GBKT), and that the majority of myeloid-dominant and T-cell-dominant clones at m12 were stable with regard to their lineage output up to m48 (Figure 7B; supplemental Figure 17D). The stability of these 3 main HSPC subsets was further confirmed by exploring the changes in the most prevalent clones over the 4 available points (Figure 7C). This analysis further supported the existence of various HSPC subsets, as defined by distinct lineage outputs with stable properties.
Discussion
In the present study, we exploited cell marking by vector integration during human gene therapy to analyze human HSPC function. CD34+ HSPCs were gene-corrected ex vivo and then transplanted into patients; this enabled us to recover, sort, and characterize vector-marked cells from peripheral blood cell lineages. Our results highlighted the difficulties associated with using this type of information to track stem cell activity: differential sampling of cell lineages, sparse sampling of cell populations, and the imperfect separation of cell fractions. We therefore modeled each of these effects and reconstructed the initial cell populations. One novel aspect of our study was the analysis of hematopoiesis in 2 distinct pathophysiologic contexts, which helps to clarify possible sources of bias related to the selective advantage of the gene-corrected cell populations. We were thus able to draw several inferences about human HSPC function. Our results suggested that in at least 2 distinct genetic diseases, human hematopoiesis after gene therapy is maintained by several distinct HSPC subsets, rather than a single subset of multipotent HSPCs.
To infer human HSPC function under physiological conditions, the various cell lineages should be uniformly marked during gene therapy. This was the case for the β hemoglobinopathy patients, whose gene-corrected cells did not have a selective advantage. In patients with WAS, however, the VCN was higher in lymphoid lineages than in myeloid lineages as a result of the selective advantage of gene-corrected lymphoid cells; this finding is in line with data from a murine model.43 Hence, this selective advantage could lead to the false detection of lymphoid-biased clones in patients with WAS. Our HSPC clonal tracking approach is novel because by normalizing against the vector input, we corrected for the imbalance in marking between cell types, and thus increase the reliability of the analysis.
A focus on highly active clones enables the output lineages to be quantified with more confidence, as it circumvents the issue of sparse sampling. This advantage is further emphasized by the results of our simulation, showing that data recovered from highly active clones should closely parallel the true blood HSPC output using our sampling scheme. However, our approach does not address the question of lineage relationships in less abundant clones. Our results also emphasize the value of analyzing later points, as ISs detected more than 2 years after cell infusion are thought to represent HSPCs capable of long-term repopulation.28
Our analysis of abundant long-term HSPCs suggested the existence of several human HSPC subsets with distinct lineage outputs. We detected multipotent HSPCs (GMBKT and GMBT clusters) accounting for about two-thirds of the total number of clones in the 4 patients analyzed. We also observed myeloid-dominant clones (accounting for an average of 14% of the clones) in 3 of 4 patients studied here, and thus in the context of 2 different types of disease. Lymphoid-dominant clones (T-cell-dominant clones, more specifically) accounted for more than 10% of the clones, even in the absence of a selective advantage in patients with a β hemoglobinopathy. The restricted clones’ stability over time suggests that they correspond to intrinsically defined HSPC subsets. However, the true dynamics of the T-cell-dominant subset are more difficult to assess, given the long half-life of T cells. Thus, our finding of biased clonal output in humans mirrors the HSC heterogeneity reported in mice.5,11 This conclusion is also supported by the long-term results of 2 clinical trials showing that cells with a limited lineage potential (T-cell-dominant and myeloid-dominant cells in both trials) have long-term self-renewal and proliferation capacities.44
The existence of lymphoid-biased progenitors with self-renewal capacity in humans has been previously suggested by a comprehensive IS study of peripheral clones and BM HSPC subsets in another WAS gene therapy trial; independent ISs were detected in multilymphoid progenitors and HSCs.29 By applying a sonication-based IS analysis and error correction, our novel pipeline enabled us to characterize the clonal lineage output through a quantitative measure of HSPC clone size. This approach confirmed the existence of myeloid-biased HSPCs in humans and identified T-cell-dominant HSPC subsets for the first time. The existence of the latter subsets agrees with the results of a study of mixed chimerism after HSCT transplantation in patients with sickle cell disease.45 The researchers demonstrated that T-cell chimerism was not correlated with myeloid or B-cell chimerism, and that donor engraftment was usually lower for T cells than for other lineages. These results suggest that T-cell reconstitution might be driven by an independent HSPC subset.45
However, one can question whether the heterogeneous lineage output in fact reflects abnormal emergency hematopoiesis driven by the transplantation conditioning regimen. A recent study showed that in the absence of irradiation, HSC clones contributed homogeneously to myeloid and B-cell lineages, whereas conditioning resulted in lineage bias after transplantation.46 In contrast, other studies of steady-state hematopoiesis have also suggested the existence of distinct HSPC lineage outputs.14,15,47,48 Thus, on the basis of the present work and reports from other researchers, it appears that HSPC lineage output heterogeneity is a part of normal hematopoiesis and is modulated by various intrinsic and environmental factors.
Tracking ISs in gene therapy patients also allowed us to estimate the total number of active HSPCs. Our estimate (from 2000 to 50 000 clones) was similar to the values of 2000 to 12 000 derived from WAS and metachromatic leukodystrophy trials.26,27 This estimate suggests that fewer than 0.01% of the total corrected CD34+ cells infused were active, repopulating clones; this is more than 10 times lower than the frequency of potentially engrafting clones estimated from phenotyping with known human HSC markers (CD34+CD38−CD45RA−CD90+CD49f+).22,49 The ex vivo culture probably contribute to reduce the number of long-term repopulating clones, and it is very likely that an additional fraction of HSPC clones (perhaps 30% of the total) is quiescent and thus cannot be detected.50,51 However, we cannot exclude some remaining heterogeneity in the CD34+CD38−CD45RA−CD90+CD49f+ HSC population, and the results of other clonal tracking approaches have also suggested that only a small number of active HSPCs contribute to the homeostasis of human hematopoiesis. In an analysis of the age-related change in the X-chromosome inactivation ratio, the steady state number of HSPC clones was estimated to be around 1200.52 More recently, HSPC clonal dynamics were reconstructed with high resolution by tracking somatic mutations identified in BM HSPCs and in their mature peripheral blood progeny; the number of active HSPCs in a healthy donor was estimated to range from 50 000 to 200 000.53 This result is in line with the minimum estimate of 50 000 clones in our β0/βE patient engrafted with the highest cell dose (13.6 × 106 cells/kg). The comparison of gene therapy/transplantation settings with human steady state hematopoiesis (using our present approach and analysis of somatic mutations53,54 ) would be a valuable way of further optimizing gene therapy protocols.
Our HSPC clonal tracking pipeline constitutes a robust platform for comparing different trials and assessing the effect of various factors on long-term hematopoietic reconstitution. In the present study, the number of highly active clone was lower in the β0/βE patient than in the other 3 patients; this might have been a result of the high total number of clones detected or the distinct HSPC source. In both autologous and allogeneic transplantation protocols, cells sourced from MPB have largely replaced those sourced from BM.55,56 Mobilization using granulocyte colony-stimulating factor (either alone or combined with Plerixafor) increases the CD34+ stem cell content and accelerates the restoration of blood cell counts,57 relative to BM-sourced HSPCs, perhaps as a result of the larger number of progenitors. Many studies have shown that MPB-sourced HSPCs (mobilized with granulocyte colony-stimulating factor alone, plerixafor alone, or a combination of the 2) and BM-sourced HSPCs display intrinsic differences in their long-term multipotency and cell cycle characteristics.33,58,59 Further longitudinal follow-up and the analysis of additional patients will be needed to assess putative intrinsic differences in stem cell sources and their effect on the long-term lineage output. Ex vivo culture is another key parameter that might modify HSPC function and lineage output.60-62 Recent research has suggested that HSPC engineering might benefit from shorter culture times or the use of compounds that stimulate HSPC self-renewal.63,64
In summary, the present study introduced a rigorous statistical approach for tackling the technical challenges associated with the use of IS data for HSPC tracking. Our results emphasized the heterogeneity of human HSPCs. Looking ahead, the long-term follow-up of gene therapy patients will facilitate the characterization of HSPC subset dynamics and the investigation of hematopoietic hierarchies in humans. Ultimately, this type of assessment might help optimize the isolation and handling of HSPCs for gene therapy and transplantation.
The full code listing and the set of postprocessed data used for the analysis and the figures are available online at https://github.com/BushmanLab/HSC_diversity. All the sequence data used in the present study are available in the NCBI Sequence Research Archive (SRA, reference: SRP139090).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Charles Berry for pivotal guidance during the early phases of this work. The authors also thank Bluebird bio for support with the β hemoglobinopathy trial (NCT 02151526) and Généthon for sponsoring the Wiskott-Aldrich syndrome trial (NCT02333760). The authors are grateful to Olivier Pellé at the Necker Cytometry Facility for assistance with cell sorting. They acknowledge Rawya Zreik for help with statistical analysis. The authors are grateful to members of the F.D.B. laboratory for help and advice.
This work was funded by National Institutes of Health grants AI 052845-13, AI 082020-05A1, AI 045008-15, U19AI117950-01, UMIAI126620 and the Penn Center for AIDS Research to F.D.B., the European Research Council (ERC Regenerative Therapy 269037 and Gene for Cure 693762) and the Dior chair for tailored medicine to M.C. This work was also funded by The Wellcome Trust (090233/Z/09/Z) and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust, and University College London (A.J.T.).
Authorship
Contribution: E.S., I.A., M.C., and F.D.B. designed the experiments; A. Guilloux, A.D., A.L., R.V., and N.C. designed and performed the statistical analysis; A.M., M.D., E.M., S.H.-B.-A., A. Galy, A.F., A.J.T., and M.C. designed and conducted the clinical trials; L.C., C.R., C.P., and S.S. performed and analyzed the experiments; F.M., J.G., C.L.N., J.K.E., and F.D.B. sequenced and analyzed the samples; E.S., A. Guilloux, M.C., and F.D.B. wrote the article; and all authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emmanuelle Six, Imagine Institute, Laboratory of Human Lymphohematopoiesis, 24 Boulevard du Montparnasse, 75015 Paris, France; e-mail: emmanuelle.six@inserm.fr; and Frederic D. Bushman, Department of Microbiology, Perelman School of Medicine at the University of Pennsylvania, 425 Johnson Pavilion, 3610 Hamilton Walk, Philadelphia, PA 19104-6076; e-mail: bushman@pennmedicine.upenn.edu.