Key Points
BTKi therapy achieves durable disease control for patients with CLL after progression on venetoclax.
Prior remission duration ≥24 months and deep response on venetoclax are associated with longer PFS on subsequent BTKi therapy.

Abstract
Highly active BTK inhibitors (BTKis) and the BCL2 inhibitor venetoclax have transformed the therapeutic landscape for chronic lymphocytic leukemia (CLL). Results of prospective clinical trials demonstrate the efficacy of venetoclax to salvage patients with disease progression on BTKis, but data on BTKi therapy after disease progression on venetoclax are limited, especially regarding durability of benefit. We retrospectively evaluated the records of 23 consecutive patients with relapsed/refractory CLL who received a BTKi (ibrutinib, n = 21; zanubrutinib, n = 2) after stopping venetoclax because of progressive disease. Median progression-free survival (PFS) and median overall survival after BTKi initiation were 34 months (range, <1 to 49) and 42 months (range, 2-49), respectively. Prior remission duration ≥24 months and attainment of complete remission or undetectable measurable residual disease on venetoclax were associated with longer PFS after BTKi salvage (P = .044 and P = .029, respectively). BTKi therapy achieved durable benefit for patients with the BCL2 Gly101Val venetoclax resistance mutation (estimated 24-month PFS, 69%). At a median survivor follow-up of 33 months (range, 2-53), 11 patients remained on BTKi and 12 had stopped therapy because of disease progression (n = 8) or toxicity (n = 4). Our findings indicate that BTKi therapy can provide durable CLL control after disease progression on venetoclax.
Introduction
The BCL2 inhibitor venetoclax1 is effective in treating relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL), including disease harboring traditional adverse prognostic factors.2-4 Venetoclax-based therapy frequently induces deep remissions, including undetectable measurable residual disease (uMRD),5 but progressive disease (PD) is common with longer-term follow-up.6 Mechanisms of venetoclax resistance include acquisition of BCL2 mutations (eg, Gly101Val7 ) and upregulation of other prosurvival proteins through microenvironment signaling, which may be interrupted by B-cell receptor inhibitors.8
The BTK inhibitor (BTKi) ibrutinib achieves durable disease control in treatment-naïve and R/R CLL, including in high-risk subgroups.9,10 Acalabrutinib (ACP-196) and zanubrutinib (BGB-3111) are next-generation BTKis with greater selectivity for BTK and similarly high response rates.11,12 Although prospective studies have demonstrated the efficacy of venetoclax postibrutinib,13 limited data exist to inform choice of therapy postvenetoclax.14-18 Here, we report the characteristics and outcomes of patients with R/R CLL who received BTKi after venetoclax discontinuation for PD.
Methods
Study design
We retrospectively reviewed data from 23 consecutive patients with R/R CLL, treated at the Royal Melbourne Hospital and Peter MacCallum Cancer Centre from September 2011 to September 2019, who received BTKi therapy for CLL after disease progression on venetoclax within 1 of 4 trials (registered at www.clinicaltrials.gov as #NCT01328626,2 #NCT01682616,4 #NCT01889186,3 and #NCT0298073119 ; supplemental Table 1, available on the Blood Web site). These patients had been treated with venetoclax at 100 to 600 mg per day (majority, 400 mg per day), with 7 receiving concurrent rituximab. Disease progression occurred during continuous venetoclax therapy in 22 patients and during venetoclax retreatment in 1 patient who had relapsed after interrupting therapy in complete remission (CR). Progressive CLL after venetoclax was treated with ibrutinib at 420 mg per day (n = 21) or zanubrutinib at 160 mg twice per day (n = 2).
Study patients
Pre-BTKi patient characteristics were recorded, including age, prior therapies, fludarabine refractoriness (failure to respond or PD ≤6 months after fludarabine-based therapy20 ), bulky adenopathy (nodes >5 cm on computed tomography21 ), del(17p), TP53 mutation, and complex karyotype (CK; ≥3 clonal chromosomal aberrations on conventional metaphase karyotype22,23 ). TP53 variants were detected with a sensitivity of 5% variant allele frequency using targeted next-generation sequencing of all coding exons of TP53 (NM_000546.5) and assessed for pathogenicity using curation sources including the Genome Aggregation Database, Catalogue of Somatic Mutations in Cancer, and the International Agency for Research on Cancer (IARC) TP53 Database. BCL2 Gly101Val mutation was detected using droplet digital polymerase chain reaction.7 uMRD was defined as detection of <1 CLL cell per 104 leukocytes in peripheral blood or bone marrow using multiparameter flow cytometry, analyzing >200 000 leukocytes.24
Statistical analysis
Kaplan-Meier method was used to estimate progression-free survival (PFS) and overall survival from BTKi initiation. Patients with planned interruption of BTKi treatment preceding stem cell transplantation (SCT) without prior PD were censored at BTKi cessation. Patients without an event were censored at date of last follow-up or data cutoff (30 September 2019) when last follow-up occurred later. Associations between clinicopathological variables and outcomes were analyzed using the Cox proportional hazards model to calculate hazard ratios with α = 0.05. All data were analyzed using STATA software (verison 14.1 for Mac) and GraphPad Prism (version 7.0b for Mac).
Results and discussion
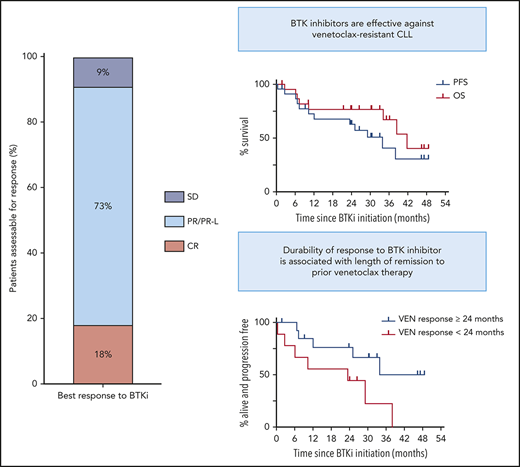
Pre-BTKi cohort characteristics are summarized in Table 1. Median age at BTKi initiation was 72 years (range, 50-89). Median time from CLL diagnosis to initiation of venetoclax and BTKi among the 19 patients for whom data were available was 8 years (range, 3-19) and 11 years (range, 5-22), respectively. Median number of prior therapies was 4 (range, 2-9); 21 patients (91%) had received fludarabine plus cyclophosphamide plus rituximab. No patient had been previously treated with B-cell receptor inhibitors. A majority of patients had disease harboring TP53 abnormalities (76%) or CK (68%) pre-BTKi. All lesions had been documented prevenetoclax except 2 new cases of TP53 abnormalities and 4 cases of CK (previously noncomplex, n = 2; previously untested, n = 2) detected after progression on venetoclax (Figure 1A).
![Outcomes of BTKi therapy for CLL after progression on venetoclax (VEN). (A) Individual patient characteristics and timelines of outcomes and treatments after BTKi initiation. Arrows at the ends of lanes indicate ongoing response to treatment at last follow-up, circles indicate death, and other treatments have specific symbols as indicated. The columns on the left indicate relevant pre-BTKi features. Gray fill indicates presence of a variable; white fill indicates absence. Where a TP53 abnormality or CK was first detected after progression on VEN, the cells have a diagonal line. In all other cases, the CLL patient had these genetic features before treatment with VEN. Patients who were treated for RT before BTKi are denoted by a red fill. Ibrutinib was the BTKi used in all patients except where zanubrutinib (ZANU) is indicated. Histology, treatment, and response of RT were as follows: lanes 1 and 5: diffuse large B-cell lymphoma (DLBCL): rituximab, ifosfamide, carboplatin, and etoposide followed by autologous (auto) SCT (CR in both cases); lane 6: DLBCL: rituximab, methotrexate, vincristine, and procarbazine followed by radiotherapy (partial remission [PR]); lane 13: Hodgkin variant: doxorubicin, bleomycin, vinblastine, and dacarbazine (CR); and lane 14: Hodgkin variant: cyclophosphamide, doxorubicin, etoposide, and prednisolone followed by radiotherapy (CR). (B) PFS (blue) and overall survival (OS) on BTKi (red) from initiation of BTKi after disease progression on VEN. In both, patient data were censored at allogeneic (allo) SCT if this occurred in first response to BTKi. (C) PFS on BTKi stratified by prior remission to VEN ≥24 (blue) or <24 months (red). HR, hazard ratio; UNK, unknown.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/25/10.1182_blood.2020004782/1/m_bloodbld2020004782f1.png?Expires=1769320652&Signature=hJGl45hfpBBkOJCDZwWFBCIVQwf1mOUhAxqUprMGo6h7eAe5KT~A8mhaojnk5shvju8hU-IBoAa-RSzaMLenpNrvvGRbqzM2a9VyXo35ahPQ80syzFh7qzCrj1mCN-VSf9bKaKhpHW~zAh1DsVlUzVVcBQk1TsZZRUPXNkX5n4k9C09ofrhnlkY7TorWCBw9gcFUDguySWByRPOKNGJZk6GsF4lc-T5BNm5zRbKRTSw8r77zB253CvKsi8aZrOR7uneEoyMp9u95lNU37tdDcsdxMyph31czRyZLXVfWSOVwi76VfwuUweWBiHCJs9rQj7cEvXEkjOSQj5xp6tRMpQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Outcomes of BTKi therapy for CLL after progression on venetoclax (VEN). (A) Individual patient characteristics and timelines of outcomes and treatments after BTKi initiation. Arrows at the ends of lanes indicate ongoing response to treatment at last follow-up, circles indicate death, and other treatments have specific symbols as indicated. The columns on the left indicate relevant pre-BTKi features. Gray fill indicates presence of a variable; white fill indicates absence. Where a TP53 abnormality or CK was first detected after progression on VEN, the cells have a diagonal line. In all other cases, the CLL patient had these genetic features before treatment with VEN. Patients who were treated for RT before BTKi are denoted by a red fill. Ibrutinib was the BTKi used in all patients except where zanubrutinib (ZANU) is indicated. Histology, treatment, and response of RT were as follows: lanes 1 and 5: diffuse large B-cell lymphoma (DLBCL): rituximab, ifosfamide, carboplatin, and etoposide followed by autologous (auto) SCT (CR in both cases); lane 6: DLBCL: rituximab, methotrexate, vincristine, and procarbazine followed by radiotherapy (partial remission [PR]); lane 13: Hodgkin variant: doxorubicin, bleomycin, vinblastine, and dacarbazine (CR); and lane 14: Hodgkin variant: cyclophosphamide, doxorubicin, etoposide, and prednisolone followed by radiotherapy (CR). (B) PFS (blue) and overall survival (OS) on BTKi (red) from initiation of BTKi after disease progression on VEN. In both, patient data were censored at allogeneic (allo) SCT if this occurred in first response to BTKi. (C) PFS on BTKi stratified by prior remission to VEN ≥24 (blue) or <24 months (red). HR, hazard ratio; UNK, unknown.
Outcomes of BTKi therapy for CLL after progression on venetoclax (VEN). (A) Individual patient characteristics and timelines of outcomes and treatments after BTKi initiation. Arrows at the ends of lanes indicate ongoing response to treatment at last follow-up, circles indicate death, and other treatments have specific symbols as indicated. The columns on the left indicate relevant pre-BTKi features. Gray fill indicates presence of a variable; white fill indicates absence. Where a TP53 abnormality or CK was first detected after progression on VEN, the cells have a diagonal line. In all other cases, the CLL patient had these genetic features before treatment with VEN. Patients who were treated for RT before BTKi are denoted by a red fill. Ibrutinib was the BTKi used in all patients except where zanubrutinib (ZANU) is indicated. Histology, treatment, and response of RT were as follows: lanes 1 and 5: diffuse large B-cell lymphoma (DLBCL): rituximab, ifosfamide, carboplatin, and etoposide followed by autologous (auto) SCT (CR in both cases); lane 6: DLBCL: rituximab, methotrexate, vincristine, and procarbazine followed by radiotherapy (partial remission [PR]); lane 13: Hodgkin variant: doxorubicin, bleomycin, vinblastine, and dacarbazine (CR); and lane 14: Hodgkin variant: cyclophosphamide, doxorubicin, etoposide, and prednisolone followed by radiotherapy (CR). (B) PFS (blue) and overall survival (OS) on BTKi (red) from initiation of BTKi after disease progression on VEN. In both, patient data were censored at allogeneic (allo) SCT if this occurred in first response to BTKi. (C) PFS on BTKi stratified by prior remission to VEN ≥24 (blue) or <24 months (red). HR, hazard ratio; UNK, unknown.
Median duration of remission to venetoclax pre-BTKi was 29 months (range, 1-59). Twenty-one patients (91%) had attained objective responses to venetoclax (PR, n = 15; CR, n = 6), with 4 (17%) achieving uMRD. All patients had ceased venetoclax because of PD (progressive CLL, n = 18; RT, n = 5 [Hodgkin variant, n = 2; diffuse large B-cell lymphoma, n = 3]). Each patient with RT was first treated with salvage chemotherapy (PR, n = 1; CR, n = 4), including autologous SCT (n = 2), and achieved durable control of transformed disease, with subsequent CLL progression triggering BTKi initiation. BCL2 Gly101Val mutation was detected at progression in 8 (42%) of 19 patients tested (Figure 1A).
Survival and subsequent treatments after BTKi initiation are shown in Figure 1A. Median interval between venetoclax cessation and BTKi initiation was 1 month (range, <1 to 11) for patients with progressive CLL and 25 months (range, 18-39) for patients initially treated for RT. All 20 patients for whom treatment indication was known met International Workshop on Chronic Lymphocytic Leukemia criteria.25 Twenty patients (91%) achieved objective responses to BTKi (PR/PR except for lymphocytosis, n = 16; CR, n = 4). All CRs met clinical International Workshop on Chronic Lymphocytic Leukemia criteria25 ; 3 of 4 were confirmed with computed tomography and bone marrow assessment. One patient achieved PR after early addition of concurrent venetoclax with insufficient time on BTKi monotherapy to evaluate response. Median PFS after BTKi initiation was 34 months (95% CI, 10.4 to undefined; range, <1 to 49; Figure 1B). At a median survivor follow-up of 33 months (range, 2-53), 11 patients (48%) remained on BTKis, including 1 who interrupted ibrutinib therapy for allogeneic SCT (Figure 1A lane 12). Twelve (52%) had stopped BTKis because of PD (n = 8 [RT, n = 2 (recurrent, n = 1; de novo, n = 1); CLL, n = 6]) or toxicity (n = 4 [sepsis, n = 1; pneumonia, n = 1; intracranial hemorrhage, n = 2 (occurring in context of myelodysplasia and severe thrombocytopenia treated with azacitidine, n = 1; acute hemorrhage into newly identified cerebral tumors of undetermined histology while on ibrutinib, n = 1)]). Median overall survival after BTKi initiation was 42 months (95% CI, 34.2 to undefined; Figure 1B). There were 8 deaths in total resulting from PD (n = 4) or toxicity (n = 4). Two patients derived clinical benefit from venetoclax-ibrutinib combination after sequentially progressing on single-agent venetoclax and then single-agent ibrutinib for 15 and 10 months, respectively (Figure 1A, lanes 6 and 7).
Univariate analyses were performed to explore putative clinicopathological variables associated with PFS after BTKi initiation (Table 1). Patients achieving remission with venetoclax ≥24 months had longer PFS on subsequent BTKi of borderline significance (median PFS for remission ≥24 vs <24 months, 34 vs 23 months; P = .044; hazard ratio, 0.31; 95% CI, 0.09-1.03; Figure 1C). All 8 patients who achieved CR/uMRD with venetoclax were in the subgroup achieving prolonged remission with venetoclax, and a similar association with prolonged PFS on BTKi was seen (P = .029). All 4 patients with prior uMRD on venetoclax remained on BTKi at a median of 21 months (range, 2-46). These univariate associations require cautious interpretation, given the modest patient numbers, and require confirmation in future analyses of larger cohorts. Among the 8 patients in whom the BCL2 Gly101Val mutation was identified, median PFS has not been reached at a median survivor follow-up of 33 months (range, 2-49; estimated 24-month PFS, 69%; supplemental Figure 1); median duration of remission with venetoclax was 37 months (range, 19-59).
In summary, these data indicate that BTKi therapy is an effective therapeutic option for patients with heavily pretreated, high-risk CLL after progression on venetoclax, including patients with prior RT in remission or CLL harboring the BCL2 Gly101Val mutation. We await further prospective trial data to validate therapeutic sequencing in the novel agent era.
Original data can be requested through the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
The authors thank all the patients, family members, and staff who participated in the studies.
Authorship
Contribution: J.F.S., A.W.R., C.S.T., and M.A.A. conceived of the project and designed the study; J.F.S., A.W.R., C.S.T., M.A.A., S.M.H., and B.J.K. were responsible for patient care; D.A.W. contributed flow cytometry and molecular data; V.S.L., T.E.L., and S.M.H. collected the data; V.S.L. and T.E.L. analyzed the data; T.N. and P.B. performed and analyzed genomic data; V.S.L. and T.E.L. wrote the first version of the manuscript; and all authors reviewed the data and contributed to critical revision of the manuscript.
Conflict-of-interest disclosure: A.W.R., M.A.A., T.E.L., and V.S.L. are employees of the Walter and Eliza Hall Institute of Medical Research, which receives milestone and royalty payments related to venetoclax. A.W.R., M.A.A., and T.E.L. are recipients of a share in royalty payments paid to the Walter and Eliza Hall Institute of Medical Research. S.M.H. has received honoraria from Gilead and nonfinancial assistance from AbbVie. C.S.T. has received honoraria and research funding from AbbVie and Janssen and honoraria from BeiGene. A.W.R. has received research funding from AbbVie, Genentech, Servier, Janssen, and BeiGene. J.F.S. receives research funding from AbbVie, Genentech, Celgene, and Janssen and is an advisory board member for and has received honoraria from AbbVie, Acerta, Celgene, Genentech, Janssen, Roche, Sunesis, and Takeda. M.A.A. has received honoraria from AbbVie and CSL Behring. T.E.L. has received honoraria from AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Mary Ann Anderson, Department of Clinical Haematology, Royal Melbourne Hospital and Peter MacCallum Cancer Centre, Melbourne, VIC, Australia; e-mail: manderson@wehi.edu.au.