

Key Points
The nuclear adapter Ldb1 is required for Lmo2 oncogene-induced thymocyte self-renewal and T-cell leukemia in a mouse model of human T-ALL.
Lmo2-induced thymocyte self-renewal is controlled by Ldb1/Lmo2-nucleated transcription complexes.

Abstract
Prolonged or enhanced expression of the proto-oncogene Lmo2 is associated with a severe form of T-cell acute lymphoblastic leukemia (T-ALL), designated early T-cell precursor ALL, which is characterized by the aberrant self-renewal and subsequent oncogenic transformation of immature thymocytes. It has been suggested that Lmo2 exerts these effects by functioning as component of a multi-subunit transcription complex that includes the ubiquitous adapter Ldb1 along with b-HLH and/or GATA family transcription factors; however, direct experimental evidence for this mechanism is lacking. In this study, we investigated the importance of Ldb1 for Lmo2-induced T-ALL by conditional deletion of Ldb1 in thymocytes in an Lmo2 transgenic mouse model of T-ALL. Our results identify a critical requirement for Ldb1 in Lmo2-induced thymocyte self-renewal and thymocyte radiation resistance and for the transition of preleukemic thymocytes to overt T-ALL. Moreover, Ldb1 was also required for acquisition of the aberrant preleukemic ETP gene expression signature in immature Lmo2 transgenic thymocytes. Co-binding of Ldb1 and Lmo2 was detected at the promoters of key upregulated T-ALL driver genes (Hhex, Lyl1, and Nfe2) in preleukemic Lmo2 transgenic thymocytes, and binding of both Ldb1 and Lmo2 at these sites was reduced following Cre-mediated deletion of Ldb1. Together, these results identify a key role for Ldb1, a nonproto-oncogene, in T-ALL and support a model in which Lmo2-induced T-ALL results from failure to downregulate Ldb1/Lmo2-nucleated transcription complexes which normally function to enforce self-renewal in bone marrow hematopoietic progenitors.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL), a neoplasm resulting from oncogenic transformation of immature T lymphocytes, frequently occurs in children and represents approximately 20% of all human ALLs.1 Activating (gain-of-function) mutations in Notch1, a signaling protein required for early stages of T-cell development, are the most common oncogenic events in T-ALLs.1 However, a subgroup of aggressive T-ALLs with poor prognosis, designated early T-cell precursor ALL (ETP-ALL) includes neoplasms that arise from a different etiology.2 ETP-ALLs, which exhibit a phenotype resembling the most immature thymocyte subsets, are frequently associated with misexpression or overexpression of the small adapter protein Lmo2.2 Evidence for a causal relationship between Lmo2 and induction of T-ALL is that transgenic overexpression of Lmo2 in immature thymocytes results in T-ALL with high penetrance in mice3-5 and that a subgroup of these neoplasms resembles human ETP-ALL.5,6 Integration-induced activation of LMO2 was also found to induce T-ALL in 25% of patients undergoing retrovirus-mediated hematopoietic stem cell gene replacement therapy for severe combined immunodeficiency (SCID-X1).7
Lmo2 contains 2 conserved cysteine rich (LIM) domains that bind to basic helix-loop-helix (bHLH) transcription factors (the hematopoietic class II proteins Tal1 and Lyl1 and the more widely expressed class I E-proteins, E2A and HEB) and to GATA family transcription factors.8-11 Lmo2 also binds to Ldb1, an adapter protein that contains a LIM interaction domain and a dimerization domain that mediates long-range promoter or enhancer interactions required for target gene expression.12-16 In hematopoietic stem/progenitor cells (HSPCs), Ldb1 and Lmo2 assemble into higher-order multiprotein complexes (designated Ldb1/Lmo2 complexes) together with the DNA binding subunits Gata2 and Tal1 (and possibly Lyl1) paired with E2A.17 Ldb1/Lmo2 complexes are required for HSPC maintenance and self-renewal, and they bind to and regulate the expression of multiple HSPC-critical genes, including Hhex, Lyl1, Gfi1b, Myb, and Nfe2.17 In addition, Ldb1/Lmo2 complexes positively autoregulate the expression of Lmo2, Lyl1, Tal1, and Gata2 in hematopoietic progenitors.17
Transgenic overexpression of Lmo2 in immature thymocytes imposes a program of long-term self-renewal resembling that observed in HSPCs.6,18 Self-renewing thymocytes in Lmo2 transgenic (Lmo2-tg) mice are radiation resistant and preclude periodic entry of bone marrow–derived lymphoid progenitors into the thymus, which results in the establishment of a pool of self-renewing intrathymic progenitors that are vulnerable to second-hit oncogenic mutations.18 It has been suggested that Lmo2-induced self-renewal and the eventual induction of T-ALL is caused by misexpression or prolonged expression of Ldb1/Lmo2-nucleated transcription complexes in immature thymocytes.6,16,19,20 Supporting this idea is the documented requirement for Ldb1 for HSPC self-renewal17 and the finding that several potential Ldb1/Lmo2 complex subunits, including Ldb1, Lmo2, Lyl1, and Gata2 are expressed in the earliest thymocyte subsets (ETPs), which phenotypically resemble HSPCs.21 However, other possible mechanisms have been proposed for Lmo2-induced T-ALL (eg, sequestration of bHLH transcription factors by Lmo2),22-25 and to date, no direct experimental evidence has been advanced supporting the Ldb1/Lmo2 transcription complex model.
In this study, we examined the role of Ldb1 in Lmo2-induced T-ALL by generating a mouse model in which Ldb1 expression could be terminated in Lmo2-tg thymocytes by Cre recombinase–mediated Ldb1 deletion. Our results identify a critical requirement for Ldb1 for Lmo2-induced thymocyte self-renewal, radiation resistance, and neoplastic transformation to T-ALL. Ldb1 was also required for acquisition of the aberrant ETP-ALL gene expression signature in immature Lmo2-tg thymocytes. Ldb1/Lmo2 complexes were detected at the promoters of key upregulated genes in Lmo2-tg thymocytes that are associated with ETP-ALL (Lyl1, Hhex, and Nfe2), and binding of both Ldb1 and Lmo2 as well as transcription of these genes was reduced by deletion of Ldb1. Collectively, these findings support a model in which Lmo2-induced self-renewal is caused by prolonged or misexpression of Ldb1/Lmo2 complexes that predispose thymocytes to oncogenic transformation resulting in T-ALL.
Methods
Detailed methods are provided in supplemental Methods, available on the Blood Web site. The RNA sequencing (RNA-seq) data set (GSE129244) has been deposited in the Gene Expression Omnibus (GEO) public database.
Results
Ldb1 is not essential for T-cell development
To determine whether Ldb1 is required for T-cell lymphopoiesis, we generated Rag1-Cre;Ldb1flox/flox (Ldb1fl/fl) mice in which expression of Cre recombinase is initiated in post-HSPC bone marrow lymphoid progenitors and continues throughout all stages of T-cell development.26 Immature CD4−CD8− (double negative [DN]) thymocytes progress through 4 stages before transitioning to the intermediate CD4+CD8+ (double positive [DP]) stage: CD25−c-kit+ (DN1/ETP), CD25+c-kit+ (DN2), CD25+c-kitlo/– (DN3), and CD25−c-kit− (DN4). To assess the timing of Cre expression, we introduced a Rosa26-EGFPf reporter which expresses EGFP only after Cre-mediated deletion of a floxed stop sequence 5′ of the EGFP complementary DNA (cDNA).27 Expression of EGFP was detected in a high percentage of all early thymocyte subsets (DN1-4) in Rosa26-EGFPf;Rag1-Cre;Ldb1fl/fl mice (Figure 1A), and deletion of the Ldb1 gene and absence of Ldb1 protein was confirmed in adult Rag1-Cre;Ldb1fl/fl thymocytes (Figure 1B-C). Because Lmo2 is normally expressed in only the most immature DN1/ETP thymocytes (which represent <0.1% of total thymocytes21 ), Lmo2 protein was undetectable in total thymocyte lysates (Figure 1C). Deletion of Ldb1 did not affect thymocyte development (Figure 1D-E). A previous analysis of Rag1-Cre;Lmo2fl/fl mice demonstrated that Lmo2 is also not required for normal T-cell development.26 Thus, although both Ldb1 and Lmo2 are each essential for HSPC maintenance and self-renewal and for erythropoiesis,17,28-30 neither protein is essential for thymopoiesis.
![Ldb1 is not essential for T-cell development. (A) Expression of the Rosa26-EGFPfCre reporter in DN subsets from Rosa26-EGFPf;Rag1-Cre;Ldb1fl/fl mice. Numbers are percent of gated cells that express EGFP. Similar subsets from non-Cre transgenic Rosa26-EGFPf mice are shown as negative control. Shown is 1 representative of 12 mice analyzed from 6 independent experiments. (B) Deletion of Ldb1 in thymocytes mediated by Rag1-Cre. Total thymocyte DNA from the indicated mice was used as a template for polymerase chain reaction (PCR) with primers that amplify the Ldb1+ (wt), Ldb1flox (Ldb1fl), or Ldb1Δ (deleted) alleles. (C) Western blot of total thymocyte lysates from the indicated mice with anti-Ldb1, anti-Lmo2, or anti-actin (loading control). (D) Phenotype of Rag1-Cre;Ldb1fl/fl mice. Upper panels: Flow cytometry (fluorescence-activated cell sorting [FACS]) analysis of total thymocytes (Thy) from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg thymocytes consisting of immature DN cells stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN(1-4) subsets. (E) FACS analysis of total lymph node (LN) cells from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Numbers in quadrants or gates are percentage of total cells. Results shown in panels B-E are 1 representative of 3 experiments.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/25/10.1182_blood.2019000794/1/m_bloodbld2019000794f1.png?Expires=1770504430&Signature=oSLA2OkefUVA~adIz2n19Yk4l2PkmnBjw-iPBiiH3Cq6l4AfDZmEc1240JHAmyrT~Dtn9~D6yWmZaOw0gOuuu9UOMq7bnpAIWWbF34ILyqTH5P3lfcVxLFMjSMaVDBZnr77c5RVfP3P3Pdg-ROnCr3ZSlkFQxNwz2vD92gq1vHT483K0dTYEhzdDt9huT1gWXj56XTo04ml8AS5BUIYyHwQC3qEA2q9ee6oxv4GrItXHoipzy7Mf-1x6-Quuz~CU13A~n-xPjEOn6r3APdqbJrvlmRUe1j-WFyry~MnbDrHVcE5ZSU-3Nb0vNFaY87FX3Vl9SYh3qIPzStASN3mOfw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ldb1 is not essential for T-cell development. (A) Expression of the Rosa26-EGFPfCre reporter in DN subsets from Rosa26-EGFPf;Rag1-Cre;Ldb1fl/fl mice. Numbers are percent of gated cells that express EGFP. Similar subsets from non-Cre transgenic Rosa26-EGFPf mice are shown as negative control. Shown is 1 representative of 12 mice analyzed from 6 independent experiments. (B) Deletion of Ldb1 in thymocytes mediated by Rag1-Cre. Total thymocyte DNA from the indicated mice was used as a template for polymerase chain reaction (PCR) with primers that amplify the Ldb1+ (wt), Ldb1flox (Ldb1fl), or Ldb1Δ (deleted) alleles. (C) Western blot of total thymocyte lysates from the indicated mice with anti-Ldb1, anti-Lmo2, or anti-actin (loading control). (D) Phenotype of Rag1-Cre;Ldb1fl/fl mice. Upper panels: Flow cytometry (fluorescence-activated cell sorting [FACS]) analysis of total thymocytes (Thy) from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg thymocytes consisting of immature DN cells stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN(1-4) subsets. (E) FACS analysis of total lymph node (LN) cells from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Numbers in quadrants or gates are percentage of total cells. Results shown in panels B-E are 1 representative of 3 experiments.
Ldb1 is not essential for T-cell development. (A) Expression of the Rosa26-EGFPfCre reporter in DN subsets from Rosa26-EGFPf;Rag1-Cre;Ldb1fl/fl mice. Numbers are percent of gated cells that express EGFP. Similar subsets from non-Cre transgenic Rosa26-EGFPf mice are shown as negative control. Shown is 1 representative of 12 mice analyzed from 6 independent experiments. (B) Deletion of Ldb1 in thymocytes mediated by Rag1-Cre. Total thymocyte DNA from the indicated mice was used as a template for polymerase chain reaction (PCR) with primers that amplify the Ldb1+ (wt), Ldb1flox (Ldb1fl), or Ldb1Δ (deleted) alleles. (C) Western blot of total thymocyte lysates from the indicated mice with anti-Ldb1, anti-Lmo2, or anti-actin (loading control). (D) Phenotype of Rag1-Cre;Ldb1fl/fl mice. Upper panels: Flow cytometry (fluorescence-activated cell sorting [FACS]) analysis of total thymocytes (Thy) from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg thymocytes consisting of immature DN cells stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN(1-4) subsets. (E) FACS analysis of total lymph node (LN) cells from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Numbers in quadrants or gates are percentage of total cells. Results shown in panels B-E are 1 representative of 3 experiments.
Ldb1 is required for induction of T-ALL in Lmo2 transgenic mice
To investigate whether Ldb1 is required for Lmo2-induced T-ALL, we generated Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice by mating Rag1-Cre;Ldb1fl/fl mice with a previously described Lmo2-tg line that overexpresses Lmo2 in all thymocytes and that develops T-ALL with long latency but high penetrance.5 Long-term monitoring of Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice revealed a significant reduction in the incidence of T-ALL compared with Ldb1fl/fl;Lmo2-tg controls (Figure 2A). One hundred percent (20 of 20) of Ldb1fl/fl;Lmo2-tg mice, but only 20% (4 of 20) of Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice developed leukemia and expired by day 435 (Figure 2A). The remaining 16 Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice showed no signs of illness, wasting, or distress.
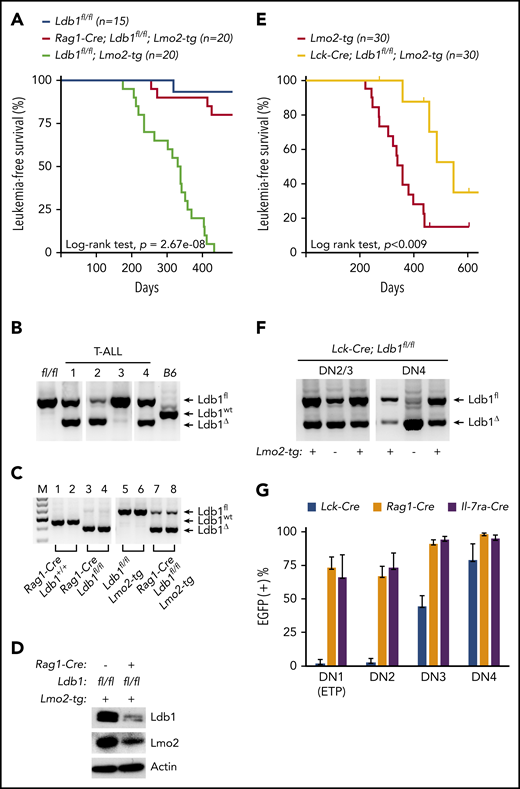
Ldb1 is required for induction of T-ALL in Lmo2-tg mice. (A) Kaplan-Meier survival plot of Ldb1fl/fl, Ldb1fl/fl;Lmo2-tg, and Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. (B) Ldb1 gene deletion in thymocytes from the 4 Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice that developed T-ALL. (C) Ldb1 gene deletion in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes. Total thymocyte DNA from the indicated mice was used as a template for PCR with primers that amplify the Ldb1+ (wt), Ldb1fl (floxed), or Ldb1Δ (deleted) alleles. M, molecular weight standard. (D) Western blot of total thymocyte lysates from the indicated mice with anti-Ldb1, anti-Lmo2, or anti-actin (loading control). (E) Kaplan-Meier survival plot of Lmo2-tg and Lck-Cre;Ldb1fl/fl;Lmo2-tg mice. (F) Ldb1 gene deletion in sorted DN2/3 or DN4 thymocytes from Lck-Cre;Ldb1fl/fl or Lck-Cre;Ldb1fl/fl;Lmo2-tg thymocytes. (G) Summary of Rosa26-EGFPf Cre reporter expression in DN1-4 subsets from Rosa26-EGFPf;Lck-Cre;Ldb1fl/fl (n = 10), Rosa26-EGFPf;Rag1-Cre;Ldb1fl/fl, (n = 12), and Rosa26-EGFPf;Il-7ra-Cre;Ldb1fl/fl (n = 9) mice. Bar graphs show mean and standard deviation of EGFP expression. Panels B, C, and F are 1 representative of 3 experiments.
Ldb1 is required for induction of T-ALL in Lmo2-tg mice. (A) Kaplan-Meier survival plot of Ldb1fl/fl, Ldb1fl/fl;Lmo2-tg, and Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. (B) Ldb1 gene deletion in thymocytes from the 4 Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice that developed T-ALL. (C) Ldb1 gene deletion in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes. Total thymocyte DNA from the indicated mice was used as a template for PCR with primers that amplify the Ldb1+ (wt), Ldb1fl (floxed), or Ldb1Δ (deleted) alleles. M, molecular weight standard. (D) Western blot of total thymocyte lysates from the indicated mice with anti-Ldb1, anti-Lmo2, or anti-actin (loading control). (E) Kaplan-Meier survival plot of Lmo2-tg and Lck-Cre;Ldb1fl/fl;Lmo2-tg mice. (F) Ldb1 gene deletion in sorted DN2/3 or DN4 thymocytes from Lck-Cre;Ldb1fl/fl or Lck-Cre;Ldb1fl/fl;Lmo2-tg thymocytes. (G) Summary of Rosa26-EGFPf Cre reporter expression in DN1-4 subsets from Rosa26-EGFPf;Lck-Cre;Ldb1fl/fl (n = 10), Rosa26-EGFPf;Rag1-Cre;Ldb1fl/fl, (n = 12), and Rosa26-EGFPf;Il-7ra-Cre;Ldb1fl/fl (n = 9) mice. Bar graphs show mean and standard deviation of EGFP expression. Panels B, C, and F are 1 representative of 3 experiments.
Because 20% of Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice developed leukemia, we examined the deletion efficiency of Ldb1 in the 4 tumor samples obtained from Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. Notably, deletion of Ldb1 was incomplete in all 4 of the T-ALL tumor samples (Figure 2B), suggesting that Ldb1 is required for Lmo2-induced oncogenesis. In contrast to non-Lmo2-tg Rag1-Cre;Ldb1fl/fl mice in which deletion of Ldb1 was essentially complete (Figure 1B), deletion of Ldb1 was incomplete in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 2C). Whereas Ldb1 protein was absent in Rag1-Cre;Ldb1fl/fltotal thymocytes (Figure 1C), Ldb1 was consistently detected in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocyte lysates, albeit at diminished levels compared with Ldb1+/+Lmo2-tg thymocytes (Figure 2D). Incomplete deletion of Ldb1 was also observed in thymocytes from Rag1-Cre;Ldb1fl/Δ;Lmo2-tg mice, which contained 1 germline deleted Ldb1 allele (Δ; data not shown) and in either Ldb1fl/fl;Lmo2-tg or Ldb1fl/Δ;Lmo2-tg mice expressing a different Cre transgene (Il-7ra-Cre) which also initiates expression in bone marrow prethymic progenitors31 (supplemental Figure 1A).
We also evaluated T-ALL frequency in Ldb1fl/fl;Lmo2-tg mice in which Ldb1 deletion was mediated by the Lck-Cre transgene which begins to be expressed in immature thymocytes but is not expressed in prethymic hematopoietic progenitors.32,33 In contrast to Rag1-Cre;Ldb1fl/fl;Lmo2-tg and Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg mice, survival of Lck-Cre;Ldb1fl/fl;Lmo2-tg mice was only modestly (although significantly) better than that of control Ldb1fl/fl;Lmo2-tg mice (Figure 2E). The reduced incidence of T-ALL in Lck-Cre;Lmo2-tg mice was not the result of a nonspecific effect of the Lck-Cre transgene because T-ALL incidence was similar in Lck-Cre;Lmo2-tg and Lmo2-tg mice (supplemental Figure 2). We also verified that neither the Lck-Cre nor the Rag1-Cre transgene altered the Lmo2-tg thymocyte phenotype independent of deletion of Ldb1 (supplemental Figure 3).
Previous results have shown that in Lmo2-tg mice, DN3 thymocytes are the critical thymocyte subset predisposing to T-ALL because only DN3 cells are capable of self-renewal and repopulation of the thymus of irradiated recipient mice after intravenous transfer.18 Analysis of Ldb1 deletion efficiency in Lck-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 and DN4 thymocytes revealed a prominent Ldb1fl band indicating inefficient deletion at the critical DN3 stage (Figure 2F). To evaluate Cre recombinase activity in all immature DN subsets, we next compared EGFP Cre-reporter expression in Lck-Cre;Ldb1fl/fl, Rag1-Cre;Ldb1fl/f, and Il-7ra-Cre;Ldb1fl/fl mice that contained the Rosa26-EGFPf allele. A high percentage of DN1/ETP, DN2, and DN3 thymocytes in Rosa26-EGFPf;Rag1-Cre Ldb1fl/f and Rosa26-EGFPf;Il-7ra-Cre;Ldb1fl/fl thymocytes were EGFP+ (Figure 2G). In contrast, <10% of DN1/ETP and DN2 thymocytes and <50% of DN3 thymocytes in Rosa26-EGFPf;Lck-Cre;Ldb1fl/fl were EGFP+ (Figure 2G). Similar patterns of EGFP expression were observed in the corresponding Lmo2-tg lines (supplemental Figure 4). However, in all 3 Cre transgenic Ldb1fl/fl lines, Ldb1 gene deletion was more complete in non-Lmo2-tg thymocytes than in the corresponding Lmo2-tg thymocytes (Figure 2B,F; supplemental Figure 1A), indicating that there is strong selective pressure against deletion of Ldb1 in Lmo2-tg DN thymocytes.
Deletion of Ldb1 restores a normal phenotype to Lmo2-tg DN thymocytes
Preleukemic Lmo2-tg DN3 thymocytes expand and accumulate in the thymus and exhibit several phenotypic abnormalities.18,34 DN cell numbers were normal in Rag1-Cre;Ldb1fl/fl;Lmo2-tg and ll-7ra-Cre;Ldb1fl/fl;Lmo2-tg mice but were elevated in Lck-Cre;Ldb1fl/fl;Lmo2-tg mice, similar to Lmo2-tg mice (Figure 3A-B; supplemental Figure 1B). The hematopoietic HSPC surface marker c-kit, which is normally downregulated in wild-type (wt) DN3 thymocytes, is highly expressed on Lmo2-tg DN3 thymocytes.18,35 Lck-Cre;Ldb1fl/fl;Lmo2-tg DN3 thymocytes but not Rag1-Cre;Ldb1fl/fl;Lmo2-tg and Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg DN3 thymocytes were c-kithi, similar to Lmo2-tg DN3 thymocytes (Figure 3A-C; supplemental Figure 1C). Lmo2-tg DN3 thymocytes also express elevated surface levels of CD93, CD5, and CD27 and reduced surface levels of CD25.18 Expression of each of these markers was partially or completely corrected on Rag1-Cre;Ldb1fl/fl;Lmo2-tg and Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg DN3 thymocytes but not on Lck-Cre;Ldb1fl/fl;Lmo2-tg DN3 thymocytes (Figure 3C; supplemental Figure 1C-D; data not shown). The DN3-DN4 developmental block caused by Lmo2 overexpression was also alleviated in both Rag1-Cre;Ldb1fl/fl;Lmo2-tg and Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg mice (Figure 3A,D; supplemental Figure 1C) but not in Lck-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 3B). Finally, with increasing age, Lmo2-tg and Lck-Cre;Ldb1fl/fl;Lmo2-tg mice, but not Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice or Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg mice exhibited a progressive accumulation of DN thymocytes and a reduction in DP and single positive (SP) thymocyte percentages and numbers (supplemental Figure 5A-B; data not shown). Collectively, these results indicated that, in addition to preventing induction of T-ALL, deletion of Ldb1 substantially corrects the abnormal phenotype and developmental defects in Lmo2-tg DN3 thymocytes.
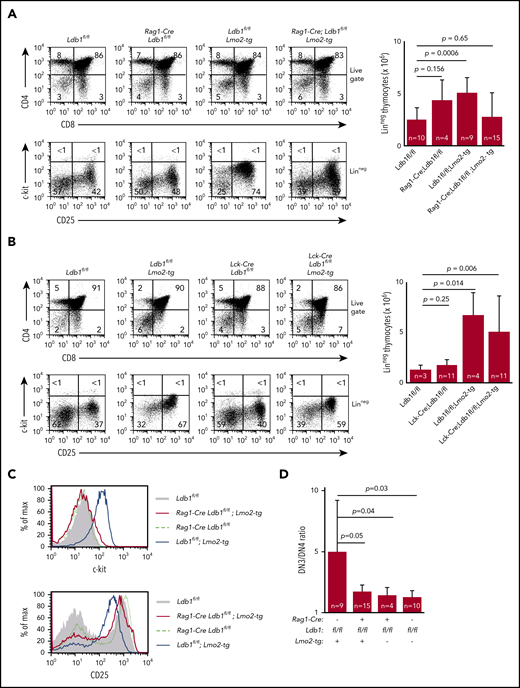
Deletion of Ldb1 reverses the abnormal phenotype of Lmo2-tg DN thymocytes. (A) Phenotype of Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. Upper panels: flow cytometry (FACS) analysis of total thymocytes from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg DN thymocytes stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN (1-4) subsets. One representative of 10 independent experiments. Right panel, numbers of linneg (DN) thymocytes from the indicated preleukemic 2- to 3-month-old mice. (B) Lck-Cre-mediated deletion of Ldb1 fails to normalize the Lmo2-tg phenotype. Upper panels: flow cytometry (FACS) analysis of total thymocytes from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg thymocytes stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN (1-4) subsets. One representative of 10 independent experiments. Right panel, numbers of linneg (DN) thymocytes from the indicated preleukemic 2- to 3-month-old mice. (C) Histograms showing surface expression of c-kit (upper) or CD25 (lower) on DN2/3 thymocytes from the indicated mice. One representative of 10 independent experiments. (D) Ratio of DN3:DN4 thymocytes in the indicated preleukemic 2- to 3-month-old mice. P values and number of mice per group are shown.
Deletion of Ldb1 reverses the abnormal phenotype of Lmo2-tg DN thymocytes. (A) Phenotype of Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. Upper panels: flow cytometry (FACS) analysis of total thymocytes from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg DN thymocytes stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN (1-4) subsets. One representative of 10 independent experiments. Right panel, numbers of linneg (DN) thymocytes from the indicated preleukemic 2- to 3-month-old mice. (B) Lck-Cre-mediated deletion of Ldb1 fails to normalize the Lmo2-tg phenotype. Upper panels: flow cytometry (FACS) analysis of total thymocytes from the indicated mice stained with fluorochrome-conjugated anti-CD4 and anti-CD8. Lower panels: gated linneg thymocytes stained with fluorochrome-conjugated anti-c-kit and anti-CD25 to delineate DN (1-4) subsets. One representative of 10 independent experiments. Right panel, numbers of linneg (DN) thymocytes from the indicated preleukemic 2- to 3-month-old mice. (C) Histograms showing surface expression of c-kit (upper) or CD25 (lower) on DN2/3 thymocytes from the indicated mice. One representative of 10 independent experiments. (D) Ratio of DN3:DN4 thymocytes in the indicated preleukemic 2- to 3-month-old mice. P values and number of mice per group are shown.
Deletion of Ldb1 reverses the abnormal self-renewal and radiation-resistance phenotype of Lmo2-tg thymocytes
After their intravenous transfer to sublethally irradiated wt (B6) mice, self-renewing Lmo2-tg DN3 thymocytes populate the recipient thymus, and within 3 to 4 weeks, they expand to occupy the entire DN compartment thus excluding entry of host bone marrow progenitors.18 In contrast, intravenously injected wt DN3 thymocytes rapidly differentiate and are replaced by host bone marrow–derived progenitors (McCormack et al18 ; data not shown). We injected thymocytes from Ldb1fl/fl;Lmo2-tg or Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice into sublethally irradiated B6 mice under experimental conditions in which donor and recipient thymocytes could be distinguished by differential expression of CD45.1 and CD45.2 (Figure 4A). Similar to previous findings,18 recipient B6 (wt) thymi were almost entirely populated by Ldb1fl/fl;Lmo2-tg donor thymocytes when analyzed 4 or 8 weeks after intravenous transfer (Figure 4B-C). In contrast, Rag1-Cre;Ldb1fl/fl;Lmo2-tg donor thymocytes were nearly undetectable in recipient thymi (Figure 4B-C), although low numbers of mature Rag1-Cre;Ldb1fl/fl;Lmo2-tg peripheral T cells confirmed that the transferred Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes had entered the thymus and differentiated (data not shown). All recipients of Lmo2-tg thymocytes, but no recipients of Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes, developed leukemia within 200 days after thymocyte transplant (Figure 4D). We confirmed that the failure of adoptively transferred Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes to repopulate the recipient thymus was the result of loss of self-renewal potential rather than a possible migration defect by performing mixed bone marrow chimera experiments (supplemental Figure 6A).
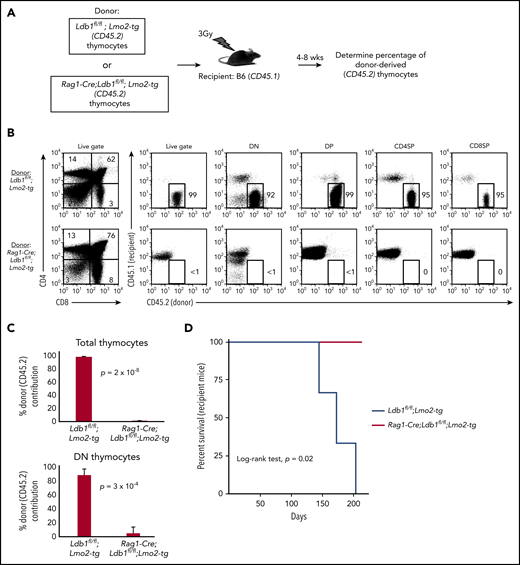
Transplantability (self-renewal potential) of Lmo2-tg thymocytes is lost following deletion of Ldb1. (A) Schematic of experimental strategy. (B) Left panels, FACS analysis of total thymocytes from recipient B6 (CD45.1) mice that had been sublethally irradiated and injected intravenously with thymocytes from an Ldb1fl/fl;Lmo2-tg (CD45.2) donor mouse (upper panel) or a Rag1-Cre;Ldb1fl/fl;Lmo2-tg (CD45.2) donor mouse (lower panel) 8 weeks before analysis. Right panels, CD45.1 vs CD45.2 staining of total (live gate) or gated DN, DP, CD4 single positive (CD4SP), or CD8SP thymocytes from the indicated recipient mice. Numbers are percentage of donor (CD45.2) thymocytes in each group. One representative of 3 experiments. (C) Percent thymocyte chimerism (total thymocytes, top; DN thymocytes, bottom) in recipient mice 8 weeks after thymocyte transfer. (D) Kaplan-Meier survival plot of recipient mice (n = 3 mice of each genotype).
Transplantability (self-renewal potential) of Lmo2-tg thymocytes is lost following deletion of Ldb1. (A) Schematic of experimental strategy. (B) Left panels, FACS analysis of total thymocytes from recipient B6 (CD45.1) mice that had been sublethally irradiated and injected intravenously with thymocytes from an Ldb1fl/fl;Lmo2-tg (CD45.2) donor mouse (upper panel) or a Rag1-Cre;Ldb1fl/fl;Lmo2-tg (CD45.2) donor mouse (lower panel) 8 weeks before analysis. Right panels, CD45.1 vs CD45.2 staining of total (live gate) or gated DN, DP, CD4 single positive (CD4SP), or CD8SP thymocytes from the indicated recipient mice. Numbers are percentage of donor (CD45.2) thymocytes in each group. One representative of 3 experiments. (C) Percent thymocyte chimerism (total thymocytes, top; DN thymocytes, bottom) in recipient mice 8 weeks after thymocyte transfer. (D) Kaplan-Meier survival plot of recipient mice (n = 3 mice of each genotype).
To assess thymocyte radiation resistance, we evaluated the capacity of in situ thymus-resident Rag1-Cre;Ldb1fl/fl;Lmo2-tg or control Lmo2-tg thymocytes to exclude entry and expansion of intravenously injected donor Lmo2-tg thymocytes after low-dose (3 Gy) irradiation of recipient mice. Six of 8 irradiated Lmo2-tg recipient thymi excluded entry or expansion of intravenously injected donor Lmo2-tg thymocytes whereas only 1 of 6 Rag1-Cre;Ldb1fl/fl;Lmo2-tg irradiated thymi excluded colonization and complete replacement by intravenously injected donor Lmo2-tg thymocytes (supplemental Figure 6B). In a separate set of experiments, we also found that, in most cases, unlike (Ldb1+/+) Lmo2-tg thymocytes, after sublethal irradiation (6.5Gy), in situ Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes were unable to prevent thymus seeding and colonization by wt (B6) bone marrow–derived T-cell progenitors (supplemental Figure 6C). Thus, Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes resemble wt B6 more closely than they resemble Lmo2-tg thymocytes because they lack self-renewal capability and radiation resistance.
Deletion of Ldb1 results in normalization of the gene expression signature of Lmo2-tg DN3 thymocytes
To evaluate the impact of Ldb1 deletion on gene expression in Lmo2-tg thymocytes, we performed RNA-seq on sorted B6 (Ldb1+/+), Lmo2-tg (Ldb1+/+), and Rag1-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 and DN4 thymocytes. Principal component analysis revealed a gene expression signature in both DN2/3 and DN4 Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes that was intermediate between B6 and Lmo2-tg thymocytes (Figure 5A). This partial rescue phenotype likely reflects the incomplete deletion of Ldb1 in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 2B). Although the incomplete deletion of Ldb1 precluded an evaluation of the impact of total loss of Ldb1 on gene expression, we inferred that a trending from dysregulated (up or down) in Lmo2-tg thymocytes toward that of normal B6 in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (which we termed “normalization”) indicated Ldb1 dependence. Interestingly, despite incomplete deletion of Ldb1, a high percentage (2559 [76%] of 3368) of the genes that were dysregulated in Lmo2-tg DN2/3 thymocytes were normalized in Rag1-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 thymocytes (ie, gene expression was significantly different in B6 and Lmo2-tg DN2/3 thymocytes but not significantly different in B6 and Rag1-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 thymocytes; Figure 5B). Hierarchical clustering of the 1091 upregulated and 1468 downregulated normalized genes failed to reveal distinct subsets of fully or partially rescued genes; instead, all genes exhibited a pattern of partial rescue, although the extent of rescue varied for different genes (supplemental Figure 7). In addition, the extent of rescue or normalization varied between individual Rag1-Cre;Ldb1fl/fl;Lmo2-tg samples, most likely reflecting the efficiency of Ldb1 deletion within each sample (supplemental Figure 7). Gene set enrichment analysis of RNA-seq data from DN2/3 thymocytes demonstrated that many of the Lmo2-dysregulated genes that were normalized by deletion of Ldb1 are important for immune system processes, T-cell differentiation, and T-cell activation and signaling, including Ptcra, CD3γ, CD3ε, Lck, and Rag2 as well as genes that regulate cell cycle progression and survival (Figure 5C). In general, genes that promote T-cell development were downregulated in Lmo2-tg DN2/3 thymocytes but were restored by deletion of Ldb1. Consistent with the relative quiescence of Lmo2-tg DN2/3 thymocytes, several genes known to have a positive role in cell proliferation (Lck, Bcl2, Bcl11a, Rhoh) were downregulated, whereas genes that negatively regulate cell proliferation (Cdkn2c, Gdf11, Jarid2, Tgfb1, Tgfb2, Smad3) were upregulated in Lmo2-tg DN2/3 thymocytes relative to wt B6 thymocytes (Figure 5C). Expression of key driver genes previously reported to be strongly upregulated in Lmo2-tg thymocytes, including Nfe2, Cpa3, Stat3, Stat5a/b, Hhex, and Lyl1,6,18,36,37 was normalized in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 5D-E). Genes that were strongly downregulated in Lmo2-tg thymocytes, including Cdkn1a, Tnfrsf8, and H19 were also much less dysregulated in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 5D), which reflects the trend toward normalization after deletion of Ldb1.
![Deletion of Ldb1 normalizes gene expression in Lmo2-tg DN thymocytes. (A) Principal component (PC1/2) analysis of RNA-seq gene expression data from B6 (wt, 4 replicates), Lmo2-tg (5 replicates), and Rag1-Cre;Ldb1fl/fl;Lmo2-tg (5 replicates) DN2/3 and DN4 thymocytes. (B) Venn diagrams depicting number of differentially expressed (DE) genes in the DN2/3 population comparisons. Deletion of Ldb1 lowers the number of Lmo2-induced DE genes from 3369 to 809. (C) Gene set enrichment analysis of RNA-seq data. Genes associated with immune system processes, T-cell differentiation, activation, signaling, or cell proliferation and survival are downregulated in Lmo2-tg DN2/3 thymocytes, whereas genes associated with negative regulation of cell proliferation and growth are upregulated in Lmo2-tg DN2/3 thymocytes. Expression of these genes is normalized in Rag1-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 thymocytes. (D) Genes previously reported to be up- or downregulated in Lmo2-tg DN thymocytes relative to wt (non–Lmo2-tg; B6) thymocytes are substantially normalized by deletion of Ldb1. (E) Genes previously shown to be positively regulated by Ldb1/Lmo2 complexes in hematopoietic progenitor cells17 are upregulated in Lmo2-tg DN2/3 thymocytes, and their expression is substantially normalized by deletion of Ldb1. For each of the genes shown in panels C-E, normalization of gene expression in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (ie, trending toward that of wt [B6] compared with Lmo2-tg) was statistically significant (P < .05). FDR, false discovery rate.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/25/10.1182_blood.2019000794/1/m_bloodbld2019000794f5.png?Expires=1770504430&Signature=hFRK0ao~G~jwtBUDR9FcEO7lfx2BPsV~O36Vd2gcDNOU~6gn6dOFqfm7hoNLJ7uFSL4tUYGb0b9Ppw9vSvOd16E5unfswSPaeLMq9KPAuGqaTETT0e2KttMXWg4S6G0M767OsnqA~Tg0-0TQl1tolX1QelGBF~ch4lHtvOguV8EPsaYb7-Tgtr7xOCKljaZjkKR3hPFZbQTtNPZp~-MIZcin~~2LvKK-pbFukw8pbTSbCnM3NpDTQEdwm7Q6LoGeT6rMU5RQ506AgeADhQSKmEnF0D-WiXY7P-Xx2-lNho-GnYpq22hgD2OcGjOP-jsw4yb-QtGGUPxeszjGTvRnlg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Deletion of Ldb1 normalizes gene expression in Lmo2-tg DN thymocytes. (A) Principal component (PC1/2) analysis of RNA-seq gene expression data from B6 (wt, 4 replicates), Lmo2-tg (5 replicates), and Rag1-Cre;Ldb1fl/fl;Lmo2-tg (5 replicates) DN2/3 and DN4 thymocytes. (B) Venn diagrams depicting number of differentially expressed (DE) genes in the DN2/3 population comparisons. Deletion of Ldb1 lowers the number of Lmo2-induced DE genes from 3369 to 809. (C) Gene set enrichment analysis of RNA-seq data. Genes associated with immune system processes, T-cell differentiation, activation, signaling, or cell proliferation and survival are downregulated in Lmo2-tg DN2/3 thymocytes, whereas genes associated with negative regulation of cell proliferation and growth are upregulated in Lmo2-tg DN2/3 thymocytes. Expression of these genes is normalized in Rag1-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 thymocytes. (D) Genes previously reported to be up- or downregulated in Lmo2-tg DN thymocytes relative to wt (non–Lmo2-tg; B6) thymocytes are substantially normalized by deletion of Ldb1. (E) Genes previously shown to be positively regulated by Ldb1/Lmo2 complexes in hematopoietic progenitor cells17 are upregulated in Lmo2-tg DN2/3 thymocytes, and their expression is substantially normalized by deletion of Ldb1. For each of the genes shown in panels C-E, normalization of gene expression in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (ie, trending toward that of wt [B6] compared with Lmo2-tg) was statistically significant (P < .05). FDR, false discovery rate.
Deletion of Ldb1 normalizes gene expression in Lmo2-tg DN thymocytes. (A) Principal component (PC1/2) analysis of RNA-seq gene expression data from B6 (wt, 4 replicates), Lmo2-tg (5 replicates), and Rag1-Cre;Ldb1fl/fl;Lmo2-tg (5 replicates) DN2/3 and DN4 thymocytes. (B) Venn diagrams depicting number of differentially expressed (DE) genes in the DN2/3 population comparisons. Deletion of Ldb1 lowers the number of Lmo2-induced DE genes from 3369 to 809. (C) Gene set enrichment analysis of RNA-seq data. Genes associated with immune system processes, T-cell differentiation, activation, signaling, or cell proliferation and survival are downregulated in Lmo2-tg DN2/3 thymocytes, whereas genes associated with negative regulation of cell proliferation and growth are upregulated in Lmo2-tg DN2/3 thymocytes. Expression of these genes is normalized in Rag1-Cre;Ldb1fl/fl;Lmo2-tg DN2/3 thymocytes. (D) Genes previously reported to be up- or downregulated in Lmo2-tg DN thymocytes relative to wt (non–Lmo2-tg; B6) thymocytes are substantially normalized by deletion of Ldb1. (E) Genes previously shown to be positively regulated by Ldb1/Lmo2 complexes in hematopoietic progenitor cells17 are upregulated in Lmo2-tg DN2/3 thymocytes, and their expression is substantially normalized by deletion of Ldb1. For each of the genes shown in panels C-E, normalization of gene expression in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (ie, trending toward that of wt [B6] compared with Lmo2-tg) was statistically significant (P < .05). FDR, false discovery rate.
We next compared our RNA-seq results with those from a published microarray study that evaluated gene expression in DN thymocytes from Lmo2-tg and Lyl1−/−;Lmo2-tg mice.37 Similar to the effect of Ldb1 deletion reported here, Lyl1 was found to be required for Lmo2 transgene-induced thymocyte self-renewal, radiation resistance, and progression to T-ALL.37 Most upregulated (Figure 6A) or downregulated (Figure 6B) genes in Lmo2-tg thymocytes that were rescued by deletion of Lyl1 were partially rescued in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes, consistent with the hypothesis that Ldb1, Lmo2, and Lyl1 function cooperatively within the same transcriptional complex.19
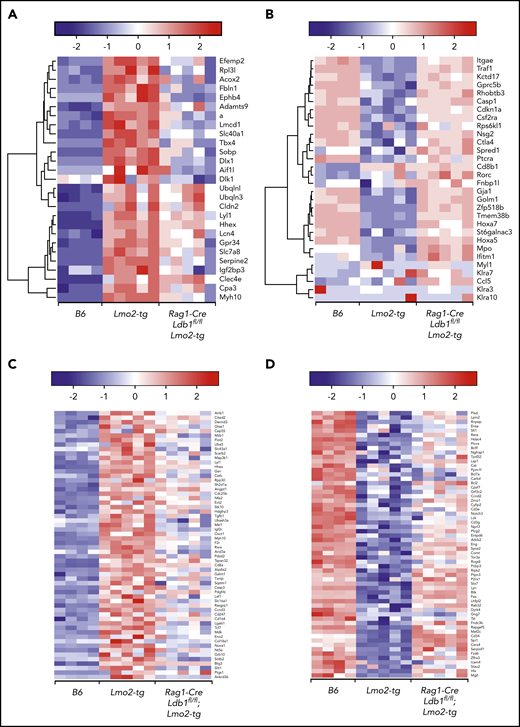
Ldb1 and Lyl1 control expression of a similar cohort of genes that are dysregulated in Lmo2-tg thymocytes. (A-B) Deletion of Ldb1 or Lyl1 has similar effects on gene expression in Lmo2-tg DN thymocytes. Shown are the expression of genes that are upregulated (A) or downregulated (B) in Lmo2-tg thymocytes and that are normalized by deletion of either Lyl137 or Ldb1. (C-D) Deletion of Ldb1 normalizes expression of genes reported to be upregulated (C) or downregulated (D) in human ETP-ALL.5,38 Results shown are from RNA-seq of DN2/3 thymocytes from B6, Lmo2-tg, or Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. For each of the genes shown in panels A-D, normalization of gene expression (ie, trending toward that of wt (B6) in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes compared with Lmo2-tg was statistically significant (P < .05).
Ldb1 and Lyl1 control expression of a similar cohort of genes that are dysregulated in Lmo2-tg thymocytes. (A-B) Deletion of Ldb1 or Lyl1 has similar effects on gene expression in Lmo2-tg DN thymocytes. Shown are the expression of genes that are upregulated (A) or downregulated (B) in Lmo2-tg thymocytes and that are normalized by deletion of either Lyl137 or Ldb1. (C-D) Deletion of Ldb1 normalizes expression of genes reported to be upregulated (C) or downregulated (D) in human ETP-ALL.5,38 Results shown are from RNA-seq of DN2/3 thymocytes from B6, Lmo2-tg, or Rag1-Cre;Ldb1fl/fl;Lmo2-tg mice. For each of the genes shown in panels A-D, normalization of gene expression (ie, trending toward that of wt (B6) in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes compared with Lmo2-tg was statistically significant (P < .05).
We also compared our results with published gene expression data from human ETP-ALL samples.5,38 Comparative analysis identified 121 differentially expressed (significantly upregulated or downregulated) genes in Lmo2-tg vs B6 DN2/3 thymocytes (false discovery rate <0.01) that were also similarly differentially expressed in human ETP-ALL vs non-ETP T-ALL (false discovery rate <0.05). Among these, 89 (73.5%) of 121 were normalized in Rag1-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 6 C-D), revealing a strong correlation in the transcriptional effects of Lmo2 overexpression in mice and human ETP-ALL and indicating that Ldb1/Lmo2 transcription complexes are important for the expression of a large number of these genes. Consistent with this interpretation, we found a high concordance of Ldb1 and Lmo2 co-expression in human T-ALL samples (supplemental Figure 8).
Finally, we analyzed the 809 dysregulated genes (623 [77%] of 809 upregulated and 186 [23%] of 809 downregulated) in Lmo2-tg thymocytes relative to B6 thymocytes that were not significantly affected by deletion of Ldb1 (supplemental Figure 9). Gene ontology analysis revealed that genes involved in signal transduction and lymphocyte differentiation (eg, ZAP-70, Aiolos, Ly9) were enriched in the upregulated gene set. The downregulated gene set contained genes that are mostly unrelated to T-cell development. In some Rag1-Cre;Ldb1f/f;Lmo2-tg samples (eg, sample 2) the expression profile trended toward that of wt (B6) thymocytes, suggesting that some of the genes included in this group may actually be Ldb1 dependent but were not identified as such by our statistical cutoff because of the variability in Ldb1 deletion efficiency among different samples. A small set of 243 genes seemed to be Ldb1 dependent but not Lmo2 dependent (ie, up- or downregulated in Rag1-Cre;Ldb1f/f;Lmo2-tg relative to B6 thymocytes but not dysregulated in Lmo2-tg thymocytes) (supplemental Figure 10). GEO profiling revealed an enrichment of genes involved in the development of nonhematopoietic tissues (muscle, urogenital, cerebellar), possibly reflecting an Lmo2-independent regulatory function for Ldb1 in the expression these genes.39
Deletion of Ldb1 reduces binding of Ldb1/Lmo2 complex(es) at key T-ALL driver genes
Together with previous data,5,16 our results suggested that Ldb1 and Lmo2 function as subunits of a transcriptional complex that includes as yet unidentified DNA binding transcription factors, and that this complex activates genes responsible for thymocyte self-renewal predisposing to T-ALL. A prediction of this model is that deletion of Ldb1 should extinguish the assembly and DNA binding of these Ldb1/Lmo2-nucleated transcription complexes in Lmo2-tg thymocytes. Although the binding sites of putative Ldb1/Lmo2-nucleated complexes have not been mapped in Lmo2-tg thymocytes, we reasoned that at least some sites may be identical to those we previously identified in HSPCs by chromatin immunoprecipitation sequencing (ChIP-seq),17 particularly sites at or near genes that are upregulated in Lmo2-tg thymocytes and human ETP-ALL and that are rescued or normalized by deletion of Ldb1. We identified binding sites for Ldb1/Lmo2 complexes in mouse HSPCs and in human ETP-ALL cell lines at the promoters of 3 T-ALL driver genes that are upregulated in Lmo2-tg thymocytes, Lyl1, Hhex, and Nfe2 (Figure 7A). Similar binding sites were confirmed in the human T-ALL cell line, LOUCY which overexpresses Lmo25 (Figure 7B) and in Lmo2-tg DN thymocytes (Figure 7A,C). ChIP quantitative polymerase chain reaction analysis confirmed the predicted reduction in Ldb1 binding at these sites in Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg thymocytes compared with Ldb1fl/fl;Lmo2-tg thymocytes (Figure 7C). Notably, Lmo2 binding was detected at the same sites and was also reduced in Il-7ra-Cre;Ldb1fl/fl;Lmo2-tg thymocytes (Figure 7D), which supports the hypothesis that Lmo2-induced dysregulation of gene expression is mediated by an Ldb1/Lmo2-nucleated transcription complex.
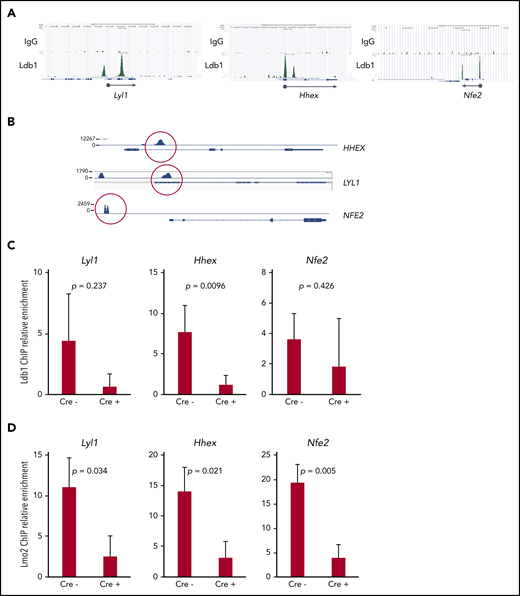
Transcription complexes that contain Ldb1 and Lmo2 bind to the promoters of key dysregulated genes in Lmo2-tg thymocytes and human T-ALL cells. (A) Binding of Ldb1 in mouse hematopoietic progenitor cells at the promoters of Lyl1, Hhex, and Nfe2 (genes that are upregulated in Lmo2-tg thymocytes). Shown are mm9 UCSC Genome Browser shots of ChIP-seq (IgG and Ldb1) performed on linneg bone marrow cells enriched for hematopoietic progenitors.17 (B) Ldb1 binding sites identified by ChIP-exo sequencing of anti-Ldb1 ChIP samples from LOUCY human T-ALL cells. (C-D) ChIP-quantitative PCR (ChIP-qPCR) analysis of Ldb1/Lmo2 complex binding sites at the Lyl1, Hhex, and Nfe2 genes (sites are shown in A). Samples for ChIP-qPCR were lineage-depleted (DN) thymocytes pooled from 3 Ldb1fl/fl;Lmo2-tg (Cre−) or 3 Il-7rα-Cre;Ldb1fl/fl;Lmo2-tg (Cre+) mice. ChIP was performed with anti-Ldb1 (C) or anti-Lmo2 (D). Bar height is the mean, and error bars show standard deviation. There was no significant difference in ChIP-qPCR results with Cre− and Cre+ samples using control primers and probes located near but outside the binding sites shown in A (data not shown).
Transcription complexes that contain Ldb1 and Lmo2 bind to the promoters of key dysregulated genes in Lmo2-tg thymocytes and human T-ALL cells. (A) Binding of Ldb1 in mouse hematopoietic progenitor cells at the promoters of Lyl1, Hhex, and Nfe2 (genes that are upregulated in Lmo2-tg thymocytes). Shown are mm9 UCSC Genome Browser shots of ChIP-seq (IgG and Ldb1) performed on linneg bone marrow cells enriched for hematopoietic progenitors.17 (B) Ldb1 binding sites identified by ChIP-exo sequencing of anti-Ldb1 ChIP samples from LOUCY human T-ALL cells. (C-D) ChIP-quantitative PCR (ChIP-qPCR) analysis of Ldb1/Lmo2 complex binding sites at the Lyl1, Hhex, and Nfe2 genes (sites are shown in A). Samples for ChIP-qPCR were lineage-depleted (DN) thymocytes pooled from 3 Ldb1fl/fl;Lmo2-tg (Cre−) or 3 Il-7rα-Cre;Ldb1fl/fl;Lmo2-tg (Cre+) mice. ChIP was performed with anti-Ldb1 (C) or anti-Lmo2 (D). Bar height is the mean, and error bars show standard deviation. There was no significant difference in ChIP-qPCR results with Cre− and Cre+ samples using control primers and probes located near but outside the binding sites shown in A (data not shown).
Massive overexpression of Lmo2 is not required for induction of T-ALL
A second model proposed for Lmo2-induced T-ALL posits that the oncogenic potential of Lmo2 is a result of its sequestration of generic bHLH proteins (E2A and/or HEB), which prevents their binding to and activation of target genes important for thymocyte maturation.22-25 Ldb1 has been shown to stabilize Lmo2.14,40 Consistent with this, we found that Lmo2 was reduced in Rag1-Cre;Ldb1f/f;Lmo2-tg thymocytes compared with Lmo2-tg thymocytes (Figure 2D), raising the possibility that the phenotypic rescue observed after deletion of Ldb1 is the result of a reduction in Lmo2 and a consequent reduction in Lmo2-mediated sequestration of E2A and HEB. To resolve this issue, we generated a Cre-inducible knockin of Lmo2 into the Rosa26 locus (supplemental Figure 11A) because our previous experience with this vector indicated that it drives relatively low expression of inserted cDNAs in thymocytes (data not shown). Lmo2 expression was induced by the Vav-iCre transgene, which is expressed in early HSPCs and in all thymocyte subsets (supplemental Figure 11B).41 Expression of (HA3 epitope tagged) Lmo2 protein was much less in Vav-iCre;Rosa26-Lmo2 DN thymocytes compared with either Lmo2-tg or Rag1-Cre;Ldb1f/f;Lmo2-tg thymocytes (supplemental Figure 11C). Despite the relatively low expression of Lmo2, similar to Lmo2-tg mice, Vav-iCre;Rosa26-Lmo2 mice developed T-ALL with long latency and at high penetrance (supplemental Figure 11D), and Vav-iCre;Rosa26-Lmo2 thymocytes were capable of populating the thymus of irradiated mice after intravenous injection (not shown). The phenotype of Vav-iCre;Rosa26-Lmo2 thymocytes also closely resembled that of Lmo2-tg thymocytes (supplemental Figure 12). Together, these data demonstrate that prolonged low-level expression of Lmo2 in DN thymocytes is sufficient to induce thymocyte self-renewal and T-ALL and that the requirement for Ldb1 for these effects is not merely to stabilize Lmo2, which argues against the sequestration model as a primary cause of Lmo2-induced T-ALL.
Discussion
In this study, we demonstrated an important function for the ubiquitous nuclear adapter and Lmo2 binding partner Ldb1 for Lmo2-induced thymocyte self-renewal and transition to T-ALL in an Lmo2-tg mouse model of ETP-ALL. Notably, Ldb1 has not been reported to be mutated, amplified, or genetically rearranged in any mouse or human cancers; thus, our results identify a key role for this nonproto-oncogene in Lmo2-induced leukemogenesis. We also provide experimental evidence that supports a model suggested by previous data5,6,17,18,36,37 proposing that a protein complex (herein designated the Ldb1/Lmo2 complex) that contains Ldb1 and Lmo2 (in addition to as yet unidentified DNA binding transcription factors) imposes an abnormal state of self-renewal on immature thymocytes and eventually results in leukemogenesis. We previously demonstrated a strict requirement for Ldb1 in the enforcement of HSPC maintenance and self-renewal.17 ChIP-seq analysis indicated that Ldb1/Lmo2 complexes bound to the promoters and/or enhancers of genes known to regulate HSPC maintenance.17 Here, we show that several of these same genes, including Lyl1, Nfe2, and Hhex, which are also upregulated in preleukemic Lmo2-tg thymocytes and in human ETP-ALL and function as key driver genes for T-ALL,5,6,18 are bound by Ldb1/Lmo2-nucleated complexes, and their expression in Lmo2-tg thymocytes is reduced and therefore normalized by deletion of Ldb1.
ETPs (DN1 thymocytes) are phenotypically similar to HSPCs, retaining multilineage differentiation potential and exhibiting an HSPC gene expression signature that includes the putative HSPC Ldb1/Lmo2 complex subunits Ldb1, Lmo2, Lyl1, Tal1, and Gata2.21 Moreover, wt thymocytes can be induced to undergo long-term self-renewal under conditions in which bone marrow progenitors are rendered noncompetitive for occupancy of intrathymic niches,42,43 and this experimentally induced thymocyte self-renewal predisposes to T-ALL.42,44,45 Thus, at the earliest DN stages, wt thymocytes retain expression of the Ldb1/Lmo2 complex subunits necessary for self-renewal, similar to HSPCs; however, with the exception of Ldb1, all of these genes are normally downregulated by the DN3 stage.21 On the basis of these and our present findings, we propose that overexpression of Lmo2 results in the continued and prolonged expression of an Ldb1/Lmo2 complex resembling, if not identical to, the HSPC Ldb1/Lmo2 complex in DN3 thymocytes. The inefficient Cre-mediated deletion of Ldb1 observed in this study suggests that the self-renewal program induced by overexpression of Lmo2 confers a strong selective advantage on DN thymocytes. In this regard, it is notable that despite intensive efforts, we were unable to delete Ldb1 in the human LOUCY T-ALL cell line, which suggests that Ldb1/Lmo2 transcription complexes may be required for the survival or self-renewal of at least some established T-ALLs (data not shown).
Another model for Lmo2-induced T-ALL which proposes that the oncogenic potential of Lmo2 is a result of its sequestration of generic bHLH proteins (E2A and/or HEB)22-25 is based on the observation that deletion of E2A and HEB results in a block in thymocyte maturation and induction of T-ALL in mice.46,47 Overexpression of the hematopoietic bHLH proteins Tal1 or Lyl1, which form heterodimers with E2A and HEB, heterozygosity of E2A or HEB,1,25 or overexpression of the E2A inhibitor Id148 accelerates induction of T-ALL. However, the recent observation that a Tal1 mutant capable of binding Lmo2 but lacking the ability to dimerize with E proteins (E2A or HEB) accelerates the onset of T-ALL in Lmo1-tg mice is inconsistent with a sequestration model.36 It is also worth noting that no evidence has been provided to indicate that deletion of E2A/HEB results in the induction of aberrant self-renewal in DN3 thymocytes similar to what is observed following misexpression or overexpression of Lmo2. In this study, we show that continued low-level expression of Lmo2 in post-ETP thymocytes is sufficient to enforce thymocyte self-renewal predisposing to T-ALL. These results suggest that, although overexpression of Lmo2 may affect the expression of some genes by sequestration of bHLH proteins, sequestration is not responsible for Lmo2-induced self-renewal and T-ALL.
ChIP-seq data suggest that Ldb1/Lmo2 complex subunits in HSPCs include Ldb1, Lmo2, Tal1, and Gata2,17 although it is likely that Lyl1 and E2A are also subunits of HSPC Ldb1/Lmo2 complexes.5,10,16,49,50 There is considerable modularity (subunit variability) in the composition of Ldb1/Lmo2 complex subunits within different hematopoietic cell populations. Whereas HSPC Ldb1/Lmo2 complexes contain Gata2, Ldb1/Lmo2 complexes in erythroid progenitors contain Gata1 in lieu of Gata2, and these complexes are responsible for transcriptional activation of most erythroid genes.17,29,30,51 Complexes containing Ldb1 and Lmo2 and either Tal1 or Lyl1 plus either Gata2 or Gata3, or complexes that contain Ldb1 and Lmo2 together with bHLH proteins but lack a GATA subunit have been identified in T-cell leukemia cell lines.5,10,16,40,52,53 A consistent finding is that Ldb1 and Lmo2 (or the closely related proteins Lmo1 or Lmo4) are present in all hematopoietic Ldb1-nucleated multiprotein complexes reported to date. The demonstrated requirement for Lyl1 but not Tal1 for both Lmo2-induced self-renewal and T-ALL in Lmo2-tg mice suggests that Lyl1 also functions as a key subunit in Lmo2 transgene-induced Ldb1/Lmo2 complexes in immature thymocytes and is necessary for progression to T-ALL.37 Identifying the precise subunit composition of Ldb1/Lmo2 complexes expressed in preleukemic Lmo2-tg thymocytes and their genomic binding sites represents an important goal for future studies that may lead to new targeted therapeutic approaches for treating ETP-ALL.
Sequence read data for ChIP-seq data shown in Figure 7 (GSE26031) has been made available in the public database.
For original data please contact LiQi Li at liliqi@mail.nih.gov.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (Project No. 1ZIAHD001803-19) (P.E.L.).
Authorship
Contribution: L.L., K.C., B.Z., S.C., J.Y.L., D.B.S., C.W., J.H., D.E.-K., and B.V. performed experiments for this study; A.M. provided bioinformatics support; L.L., K.P., U.P.D., K.Z. and P.E.L. designed experiments and edited the manuscript; and P.E.L. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paul E. Love, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Building 6B, Room 2B-210, 9000 Rockville Pike, Bethesda, MD 20892; e-mail: lovep@mail.nih.gov; and Utpal P. Davé, Division of Hematology/Oncology, Department of Medicine, Indiana University School of Medicine, Walther Hall, 980 W Walnut St, Room C312J, Indianapolis, IN 46202; e-mail: udave@iu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal