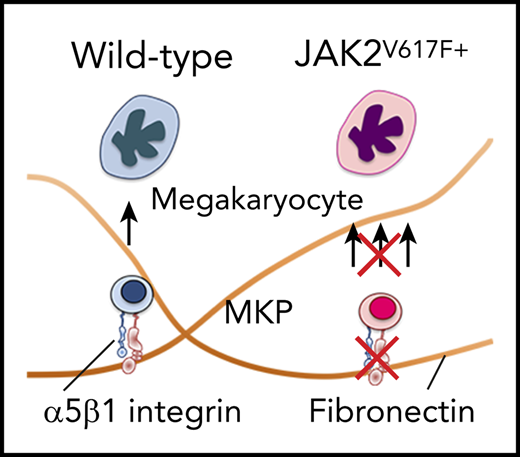
Key Points
The α5 subunit of α5β1 integrin is upregulated in JAK2V617F+ megakaryocytes in PMF leading to increased adhesion to fibronectin.
Antibody-mediated inhibition of the α5 subunit prevents expansion of megakaryocyte lineage in JAK2V617F+ cells in vitro and in vivo.

Abstract
Excessive accumulation of extracellular matrix (ECM) is a hallmark of bone marrow (BM) milieu in primary myelofibrosis (PMF). Because cells have the ability to adhere to the surrounding ECM through integrin receptors, we examined the hypothesis that an abnormal ECM-integrin receptor axis contributes to BM megakaryocytosis in JAK2V617F+ PMF. Secretion of ECM protein fibronectin (FN) by BM stromal cells from PMF patients correlates with fibrosis and disease severity. Here, we show that Vav1-hJAK2V617F transgenic mice (JAK2V617F+) have high BM FN content associated with megakaryocytosis and fibrosis. Further, megakaryocytes from JAK2V617F+ mice have increased cell surface expression of the α5 subunit of the α5β1 integrin, the major FN receptor in megakaryocytes, and augmented adhesion to FN compared with wild-type controls. Reducing adhesion to FN by an inhibitory antibody to the α5 subunit effectively reduces the percentage of CD41+ JAK2V617F+ megakaryocytes in vitro and in vivo. Corroborating our findings in mice, JAK2V617F+ megakaryocytes from patients showed elevated expression of α5 subunit, and a neutralizing antibody to α5 subunit reduced adhesion to FN and megakaryocyte number derived from CD34+ cells. Our findings reveal a previously unappreciated contribution of FN-α5β1 integrin to megakaryocytosis in JAK2V617F+ PMF.
Introduction
Primary myelofibrosis (PMF), of which JAK2V617F is the major driver mutation, is a clonal myeloproliferative disease characterized by splenomegaly, megakaryocytosis with atypical megakaryocytes, and excessive deposition of extracellular matrix (ECM) in bone marrow (BM).1-4 PMF is a condition of dismal prognosis with limited treatment options.5
BM mesenchymal stromal cells from JAK2V617F+ patients display elevated secretion of ECM protein fibronectin (FN),6,7 which correlates with severity of fibrosis.7 Adhesion to FN enhances megakaryocytic differentiation,8 and megakaryocytes produce cellular FN on their own.9 Schick et al identified α5β1 as the major FN receptor in megakaryocytes.10
Vav1-hJAK2V617F transgenic mice (JAK2V617F+)11 express the most frequent mutation in PMF and show splenomegaly, megakaryocytosis, and myelofibrosis in BM. Despite the conspicuous presence of ECM in myelofibrotic BM, the interaction of megakaryocytes with ECM and its possible contribution to disease have not been thoroughly studied in PMF. Here, using JAK2V617F+ mice and human samples, we show that a dysregulated FN-α5β1 integrin axis contributes to expansion of megakaryocyte lineage in JAK2V617F+ PMF.
Study design
JAK2V617F+ transgenic mice
JAK2V617F+ mice,11 constitutively expressing the Vav1-hJAK2V617F transgene were backcrossed with C57BL/6J, brought to homozygosity, and used within 10 generations. Age- and gender-matched C57BL/6J wild-type (WT) mice were used as controls. Protocols were approved by the Boston University Medical Campus Institutional Animal Care and Use Committee.
In vitro differentiation of megakaryocytes from BM
BM cells were cultured as previously described12 with pegylated megakaryocyte growth and development factor (25 ng/mL, PEG-MGDF). When applicable, cells were cultured on plates coated with bovine serum albumin (BSA; 50 μg/mL) or human plasma FN (50 μg/mL; Enzyme Research Laboratories) and blocked with 1% BSA/phosphate-buffered saline, with Armenian Hamster immunoglobulin G (IgG) isotype control or anti-α5 subunit antibody HMα5-1 (both at 10 μg/mL; eBiosciences). Images were acquired with Olympus IX70 microscope equipped with U-Plan FI10×/0.30 Ph1 objective, Olympus XM10 digital camera, and CellSens software (Olympus).
Histology, flow cytometry, and adhesion assay
See supplemental Methods, available on the Blood Web site.
Treatment of JAK2V617F+ mice with anti-α5 subunit antibody 5H10-27
Three doses (1 mg/kg) of rat IgG2aκ isotype control antibody (clone R35-95) or 5H10-27 (BD Biosciences) were administered via tail vein every 48 hours. Animals were analyzed 24 hours after the last dose.
Patient samples
Cells derived from five healthy controls (HC) and 7 PMF patients were used. All patients (5 males, 2 females) were JAK2V617F-mutated (4 homozygous, 3 heterozygous) and presented BM fibrosis (1 patient grade 1, 3 patients grade 2, 3 patients grade 3). Samples were obtained following guidelines of the Institutional Review Boards at the Istituto di Ricovero e Cura a Carattere Scientifico Policlinico S. Matteo Foundation, Pavia, Italy. All participants signed informed consent for study inclusion, according to rules and principles of the Helsinki Declaration.
Human megakaryocyte culture
Peripheral blood CD34+ cells were cultured in conditions that promote megakaryocyte development.13 When applicable, wells were coated with human plasma FN (100 μg/mL), and unrelated IgG2b (10 μg/mL, MCP-11; MilliporeSigma) or anti-α5 integrin antibody (10 μg/mL, SAM-1; MilliporeSigma) was added with fresh medium at days 1, 3, 7, and 10.
Results and discussion
BM histology of myelofibrotic JAK2V617F+ mice showed abundant FN in BM ECM (Figure 1A). The percentage of CD41+ and CD42d+ cells in BM, specific for total and mature megakaryocytes, respectively, were elevated as expected (Figure 1B).11
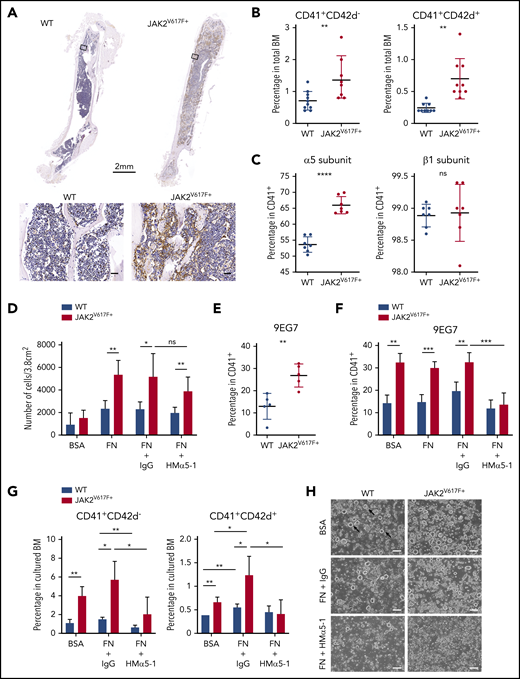
Increased expression of α5 subunit and adhesion of JAK2V617F+ megakaryocytes to fibronectin results in megakaryocytosis. (A) Immunohistochemical staining of fibronectin (brown) in BM of C57BL/6J WT and JAK2V617F+ mice (26-week-old males). Top: whole-bone scanning (×0.4 original magnification). Inset squares: approximate location of images shown in bottom panels (×20 original magnification; scale bars represent 50 μm). (B) Percentage of CD41+CD42d− and CD41+CD42d+ megakaryocytes in BM of WT (n = 6 females and 4 males, 15 weeks old) and JAK2V617F+ mice (n = 5 females and 4 males, 15 weeks old). The differential effect persisted in the 2 sexes. (C) Cell surface expression of α5 (HMα5-1 antibody) and β1 (Ha2/5 antibody) subunits on CD41+ megakaryocytes differentiated in vitro. Data represent 2 independent experiments from WT (n = 4, 21-week-old males and n = 3, 18-week-old females) and JAK2V617F+ mice (n = 3, 21-week-old males and n = 4, 18-week-old females). Note that y-axis does not start at 0. (D) Adhesion assay of in vitro-differentiated megakaryocytes on plates coated with control BSA or FN, treated with an inhibitory antibody against α5 subunit (HMα5-1) or with a hamster IgG isotype control. Data represent 2 independent experiments, from WT (n = 2, 13-week-old and n = 2, 21-week-old males) and JAK2V617F+ mice (n = 4, 13-week-old and n = 3, 21-week-old males). (E) Cell surface expression of extended conformation β1 subunit (9EG7 antibody) in CD41+ megakaryocytes differentiated in vitro from WT and JAK2V617F+ mice (n = 5 each, 14-week-old males). (F) Similar analysis as in panel E, with megakaryocytes treated with an inhibitory antibody against α5 subunit (HMα5-1) or with a hamster IgG isotype control during in vitro differentiation on BSA or FN-coated plates. Data represent WT (n = 4, 18-week-old females) and JAK2V617F+ mice (n = 3, 18-week-old females). (G) Percentage of CD41+CD42d− and CD41+CD42d+ megakaryocytes in BM cultured under conditions as in panel F. Representative data from 1 of 2 independent experiments showing similar tendency are shown, from WT (n = 3, 10-week-old females) and JAK2V617F+ (n = 4, 10-week-old females). (H) Representative bright-field images of megakaryocytes differentiated in vitro in culture conditions shown in panel G. Arrows indicate some examples of megakaryocytes (×10 original magnification; scale bars represent 100 μm). Data are expressed as mean ± standard deviation; ns, not significant. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Increased expression of α5 subunit and adhesion of JAK2V617F+ megakaryocytes to fibronectin results in megakaryocytosis. (A) Immunohistochemical staining of fibronectin (brown) in BM of C57BL/6J WT and JAK2V617F+ mice (26-week-old males). Top: whole-bone scanning (×0.4 original magnification). Inset squares: approximate location of images shown in bottom panels (×20 original magnification; scale bars represent 50 μm). (B) Percentage of CD41+CD42d− and CD41+CD42d+ megakaryocytes in BM of WT (n = 6 females and 4 males, 15 weeks old) and JAK2V617F+ mice (n = 5 females and 4 males, 15 weeks old). The differential effect persisted in the 2 sexes. (C) Cell surface expression of α5 (HMα5-1 antibody) and β1 (Ha2/5 antibody) subunits on CD41+ megakaryocytes differentiated in vitro. Data represent 2 independent experiments from WT (n = 4, 21-week-old males and n = 3, 18-week-old females) and JAK2V617F+ mice (n = 3, 21-week-old males and n = 4, 18-week-old females). Note that y-axis does not start at 0. (D) Adhesion assay of in vitro-differentiated megakaryocytes on plates coated with control BSA or FN, treated with an inhibitory antibody against α5 subunit (HMα5-1) or with a hamster IgG isotype control. Data represent 2 independent experiments, from WT (n = 2, 13-week-old and n = 2, 21-week-old males) and JAK2V617F+ mice (n = 4, 13-week-old and n = 3, 21-week-old males). (E) Cell surface expression of extended conformation β1 subunit (9EG7 antibody) in CD41+ megakaryocytes differentiated in vitro from WT and JAK2V617F+ mice (n = 5 each, 14-week-old males). (F) Similar analysis as in panel E, with megakaryocytes treated with an inhibitory antibody against α5 subunit (HMα5-1) or with a hamster IgG isotype control during in vitro differentiation on BSA or FN-coated plates. Data represent WT (n = 4, 18-week-old females) and JAK2V617F+ mice (n = 3, 18-week-old females). (G) Percentage of CD41+CD42d− and CD41+CD42d+ megakaryocytes in BM cultured under conditions as in panel F. Representative data from 1 of 2 independent experiments showing similar tendency are shown, from WT (n = 3, 10-week-old females) and JAK2V617F+ (n = 4, 10-week-old females). (H) Representative bright-field images of megakaryocytes differentiated in vitro in culture conditions shown in panel G. Arrows indicate some examples of megakaryocytes (×10 original magnification; scale bars represent 100 μm). Data are expressed as mean ± standard deviation; ns, not significant. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Analysis of α5β1 integrin cell surface expression showed significant elevation of the α5 subunit in in vitro-differentiated JAK2V617F+ megakaryocytes (Figure 1C). JAK2V617F+ megakaryocytes showed significantly higher adhesion to FN, preventable by treatment with an inhibitory antibody against the α5 subunit (HMα5-1)14 (Figure 1D). Residual adhesion could be due to incomplete inhibition by the antibody and/or adhesion through other fibronectin receptors (supplemental Figure 1A). Percentage of megakaryocytes expressing the β1 subunit did not differ in JAK2V617F+ and WT control mice (Figure 1C), but expression of extended conformation β1 subunit, a surrogate marker for integrin activation detected by the antibody clone 9EG7,15 was significantly elevated in JAK2V617F+ megakaryocytes (Figure 1E). Treatment with HMα5-1 was able to reduce 9EG7 expression, therefore integrin activation (Figure 1F). To probe for functional significance of this finding, megakaryocyte differentiation was carried out on FN-coated plates with and without presence of HMα5-1. Mature megakaryocytes were more prevalent in FN-cultured cells in both WT and JAK2V617F+ BM, underscoring the importance of FN in megakaryocytosis. Antibody-mediated inhibition of α5 subunit lowered the percentage of CD41+ and CD42d+ cells (Figure 1G-H; supplemental Figure 1B), bringing megakaryocyte percentage almost to WT levels in JAK2V617F+ BM, and increased ploidy of 8N-16N (supplemental Figure 1C) in both WT and JAK2V617F+ BM. Inverse effect on megakaryocyte number and ploidy was found curative for PMF.16
To explore the significance of the above findings in vivo, expression of α5β1 and 9EG7 was examined in fresh BM megakaryocytes and megakaryocyte progenitors (MKP) from JAK2V617F+ mice. Consistent with observations in vitro, expression of α5 subunit and 9EG7, but not of the β1 subunit, was significantly elevated in BM CD41+ megakaryocytes (Figure 2A-C; supplemental Figure 2A) and MKP (Figure 2D-G; supplemental Figure 2B) from JAK2V617F+ mice. Elevation was also detected in other fibronectin receptor subunits αIIb and β3 (supplemental Figure 2C), but not in α2 (supplemental Figure 2D).
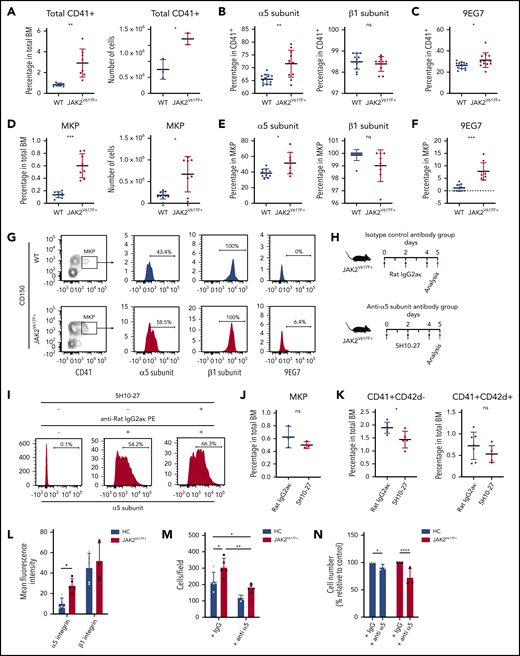
Antibody-mediated inhibition of α5 subunit decreases megakaryocyte numbers in vivo in JAK2V617F+ mice and in JAK2V617F+ patient samples. (A) Percentage and number of total CD41+ megakaryocytes in BM of WT C57BL/6J control (n = 10, 10- to 18-week-old males) and JAK2V617F+ mice (n = 11, 10- to 18-week-old males). Number of CD41+ megakaryocytes correspond to n = 3 animals from each group (14-week-old males). (B) Cell surface expression of α5 (HMα5-1) and β1 (Ha2/5) subunits on CD41+ BM megakaryocytes. Data for α5 subunit represents WT (n = 10, 10- to 18-week-old males) and JAK2V617F+ mice (n = 11, 10- to 18-week-old males); data for β1 subunit represent WT (n = 10, 12- to 14-week-old males) and JAK2V617F+ mice (n = 10, 12- to 14-week-old males). Note that y-axis does not start at 0. (C) Cell surface expression of extended conformation β1 subunit (9EG7) in CD41+ BM megakaryocytes from WT (n = 14, 18- to 25-week-old males and females) and JAK2V617F+ mice (n = 12, 18- to 25-week-old males and females). (D) Percentage out of total BM cells and number of MKP (Lin-c-Kit+Sca-1−CD41+CD150+) in BM of WT (n = 10, 19- to 25-week-old females) and JAK2V617F+ mice (n = 8, 19- to 25-week-old females). (E) Cell surface expression of α5 (HMα5-1) and β1 (Ha2/5) subunits in MKP from WT (n = 10, 19- to 25-week-old females) and JAK2V617F+ mice (n = 8, 19- to 25-week-old females). Note that y-axis does not start at 0 for β1 subunit. (F) Cell surface expression of 9EG7 in MKP from WT and JAK2V617F+ mice (n = 10 each, 18- to 21-week-old females). (A-F) Data are an aggregate of at least 3 independent experiments. (G) Representative data of flow cytometric analysis of α5 (HMα5-1), β1 (Ha2/5) subunits, and 9EG7 expression in MKP shown in panels D-F. (H) Schematic representation of 5H10-27 treatment protocol. JAK2V617F+ mice were injected with either rat IgG2aκ or 5H10-27. Black arrows indicate antibody injection; red arrows indicate date animals were analyzed. (I) Representative flow cytometric analysis of 5H10-27 antibody-labeled CD41+ cells in 5H10-27-injected animals. Cells collected from treated animals were stained ex vivo only with anti-rat IgG2aκ phycoerythrin (PE) secondary antibody (middle), or with 5H10-27 followed by anti-rat IgG2aκ PE (right). Percentage of (J) MKP and (K) CD41+ and CD42d+ cells in JAK2V617F+ animals treated with rat IgG2aκ (n = 7, 8- to 10-week-old females) or 5H10-27 (n = 5, 8- to 10-week-old females). Data represent aggregate from 3 independent experiments. (L) Mean fluorescence intensities of α5 (P1D6) and β1 (TS2/16) integrin subunits in megakaryocytes differentiated from HC and JAK2V617F+ PMF patients (n = 5 per group). (M) 2 × 105 megakaryocytes derived from HC and JAK2V617F+ PMF patients were plated on fibronectin-coated plates and incubated for 3 hours to allow for adhesion in the presence of an unrelated IgG or anti-α5 integrin antibody (SAM-1) with blocking function. Adherent megakaryocytes were counted and expressed as number of cells per field (n = 5 per group). (L-M) All 5 HC and PMF patients 1 through 5 were used in studies. (N) CD34+ cells from HC and JAK2V617F+ PMF patients were differentiated on fibronectin coated-plates in the presence of an unrelated IgG or anti-α5 integrin antibody (SAM-1) with blocking function for 13 days. Three of 5 HC and PMF patients 5 through 7 were used in studies. Normalized cell counts from 3 independent experiments are shown. Data are expressed as mean ± standard deviation; ns, not significant. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Antibody-mediated inhibition of α5 subunit decreases megakaryocyte numbers in vivo in JAK2V617F+ mice and in JAK2V617F+ patient samples. (A) Percentage and number of total CD41+ megakaryocytes in BM of WT C57BL/6J control (n = 10, 10- to 18-week-old males) and JAK2V617F+ mice (n = 11, 10- to 18-week-old males). Number of CD41+ megakaryocytes correspond to n = 3 animals from each group (14-week-old males). (B) Cell surface expression of α5 (HMα5-1) and β1 (Ha2/5) subunits on CD41+ BM megakaryocytes. Data for α5 subunit represents WT (n = 10, 10- to 18-week-old males) and JAK2V617F+ mice (n = 11, 10- to 18-week-old males); data for β1 subunit represent WT (n = 10, 12- to 14-week-old males) and JAK2V617F+ mice (n = 10, 12- to 14-week-old males). Note that y-axis does not start at 0. (C) Cell surface expression of extended conformation β1 subunit (9EG7) in CD41+ BM megakaryocytes from WT (n = 14, 18- to 25-week-old males and females) and JAK2V617F+ mice (n = 12, 18- to 25-week-old males and females). (D) Percentage out of total BM cells and number of MKP (Lin-c-Kit+Sca-1−CD41+CD150+) in BM of WT (n = 10, 19- to 25-week-old females) and JAK2V617F+ mice (n = 8, 19- to 25-week-old females). (E) Cell surface expression of α5 (HMα5-1) and β1 (Ha2/5) subunits in MKP from WT (n = 10, 19- to 25-week-old females) and JAK2V617F+ mice (n = 8, 19- to 25-week-old females). Note that y-axis does not start at 0 for β1 subunit. (F) Cell surface expression of 9EG7 in MKP from WT and JAK2V617F+ mice (n = 10 each, 18- to 21-week-old females). (A-F) Data are an aggregate of at least 3 independent experiments. (G) Representative data of flow cytometric analysis of α5 (HMα5-1), β1 (Ha2/5) subunits, and 9EG7 expression in MKP shown in panels D-F. (H) Schematic representation of 5H10-27 treatment protocol. JAK2V617F+ mice were injected with either rat IgG2aκ or 5H10-27. Black arrows indicate antibody injection; red arrows indicate date animals were analyzed. (I) Representative flow cytometric analysis of 5H10-27 antibody-labeled CD41+ cells in 5H10-27-injected animals. Cells collected from treated animals were stained ex vivo only with anti-rat IgG2aκ phycoerythrin (PE) secondary antibody (middle), or with 5H10-27 followed by anti-rat IgG2aκ PE (right). Percentage of (J) MKP and (K) CD41+ and CD42d+ cells in JAK2V617F+ animals treated with rat IgG2aκ (n = 7, 8- to 10-week-old females) or 5H10-27 (n = 5, 8- to 10-week-old females). Data represent aggregate from 3 independent experiments. (L) Mean fluorescence intensities of α5 (P1D6) and β1 (TS2/16) integrin subunits in megakaryocytes differentiated from HC and JAK2V617F+ PMF patients (n = 5 per group). (M) 2 × 105 megakaryocytes derived from HC and JAK2V617F+ PMF patients were plated on fibronectin-coated plates and incubated for 3 hours to allow for adhesion in the presence of an unrelated IgG or anti-α5 integrin antibody (SAM-1) with blocking function. Adherent megakaryocytes were counted and expressed as number of cells per field (n = 5 per group). (L-M) All 5 HC and PMF patients 1 through 5 were used in studies. (N) CD34+ cells from HC and JAK2V617F+ PMF patients were differentiated on fibronectin coated-plates in the presence of an unrelated IgG or anti-α5 integrin antibody (SAM-1) with blocking function for 13 days. Three of 5 HC and PMF patients 5 through 7 were used in studies. Normalized cell counts from 3 independent experiments are shown. Data are expressed as mean ± standard deviation; ns, not significant. *P < .05, **P < .01, ***P < .001, ****P < .0001.
We next explored the effect of inhibition of α5 subunit in vivo, a possible means of reducing megakaryocyte burden in PMF. To reduce immunogenicity, we chose the rat-derived 5H10-2717 over the hamster-derived HMα5-1 antibody. To quantify in vivo labeled cells, BM of antibody-injected animals was stained with fluorochrome-conjugated isotype-specific secondary antibody and analyzed on flow cytometry. 5H10-27 and HMα5-1 labeled the same percentage of cells in JAK2V617F+ BM, demonstrating the interchangeability of the 2 antibodies (supplemental Figure 3A). The minimal dose required to label BM cells to saturation was determined to be 1 mg/kg (supplemental Figure 3B). Labeling persisted in BM for at least 48 hours (supplemental Figure 3C).
Previous reports determined the megakaryocyte replacement time to be 60 hours.18 To allow enough time for the effect on immature megakaryocytes to become apparent in the mature population, we designed a 5-day protocol (120 hours) comprising 3 intravenous bolus injections of 5H10-27 or isotype control antibody (rat IgG2aκ) at 1 mg/kg every 48 hours (Figure 2H). Robust megakaryocyte labeling persisted up to day 5 (Figure 2I). The percentage of CD41+CD42d− megakaryocytes in 5H10-27-injected animals was significantly lower than in isotype-control antibody-injected animals. An apparent but still not significant decrease was detected in more mature CD41+CD42d+ megakaryocytes (Figure 2J-K; supplemental Figure 3D-E). Peripheral blood counts, including platelet counts, were not affected by the treatment (supplemental Table 2), likely owing to an inverse effect of 5H10-27 on megakaryocyte number and ploidy.
Consistent with mice data, human PMF megakaryocytes showed elevated levels of α5 subunit, but not β1, and a neutralizing antibody to α5 subunit reduced megakaryocyte adhesion and megakaryocyte number derived from CD34+ cells, compared with controls (Figure 2L-N; supplemental Figure 4A-C).
In sum, our results show elevated adhesion to FN in megakaryocytes from JAK2V617F+ mice and PMF patients, largely attributable to increased expression of α5 subunit of the major FN receptor α5β1 integrin.10 Relative messenger RNA expression of α5 and β1 subunits was similar between WT and JAK2V617F+ megakaryocytes (supplemental Figure 5A), suggesting that the overexpression of α5 subunit is most likely because of a posttranslational mechanism.
Inhibition of α5 subunit in vitro and in vivo resulted in lowered percentage of CD41+ megakaryocytes in JAK2V617F+ BM compared with controls. Collectively, our results suggest that strategies to target megakaryocyte α5 subunit expression and the FN receptor hold promise for therapeutic approaches in JAK2V617F+ PMF.
Contact the corresponding author for original data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Zhizhuang Joe Zhao from the University of Oklahoma for generously sharing the Vav1-hJAK2V617F transgenic mice, and Vittorio Rosti (Center for the Study of Myelofibrosis at the IRCCS Policlinico S. Matteo Foundation, Pavia, Italy) for providing patient samples. The authors also thank Kirin Brewery for sharing PEG-MGDF, Orly Leiva for BM tissue sections, and the Boston University Flow Cytometry Core Facility for equipment and technical support.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL136363) (K.R.); Associazione Italiana per la Ricerca sul Cancro (AIRC IG 2016 18700) (A.B.); Italian Ministry of Health (Ricerca Finalizzata Giovani Ricercatori GR-2016-02363136) (A.B.); National Institutes of Health, Office of Research Infrastructure Programs Special Emphasis Research Career Award (1K01_D025290-01A1) (S.M.); National Institutes of Health, National Heart, Lung, and Blood Institute Cardiovascular Research Training (T32 HL007224) (C.R.T., S.K.N., and C.M.W.); and National Institutes of Health, National Heart, Lung, and Blood Institute Research in Blood Diseases and Resources Training (T32 HL007501) (C.M.W.).
Authorship
Contribution: S.M. and K.R. generated the hypothesis; S.M. designed and performed most of experiments, analyzed data, and wrote the manuscript with K.R.’s input and editing; K.R. also reviewed data analysis; C.R.T. and S.K.N. assisted in some experiments; C.M.W. and A.K. performed quantitative reverse transcriptase polymerase chain reaction experiments; C.M. performed some adhesion assays; and A.M. and A.B. designed, and A.M. performed experiments with human cells.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Katya Ravid, Boston University School of Medicine, 700 Albany St, W-6, Boston, MA 02118; e-mail: kravid@bu.edu.